欢迎访问诺维之舟医学科研平台,我们的课程比丁香园更香!
Language: 


时间:2024-03-05 22:44:59 阅读:105

bilibili:文章对应的讲解视频在此。熊大学习社 https://space.bilibili.com/475774512
Gitee开源:https://gitee.com/shenghuaxiong/ioter
CSDN玩转物联网专栏文章:https://blog.csdn.net/shx13141/category_11669532.html
微信公众号:熊大学习社、诺维之舟
公益网站,http://nwzz.xyz/ ,可在线使用chatgpt3.5|GPT4|Midjourney

课程相关资料:
(1)学习资料,包括医学SCI绘图-基于R课程的50+种学术图片的代码和数据。关注公众号“熊大学习社”,回复“scih1214”,获取资料链接,代码在文章中。50+种学术图片的代码和数据资料在文章最下面,付费后可见。
谢谢您的支持,我们坚持学以致用、高效学习、质量服务,做好有质量的分享。
另外,服务合作请联系: sophie_sheng1122。
关注B站熊大学习社,公众号诺维之舟、熊大学习社。您的一键三连是我最大的动力。
效果
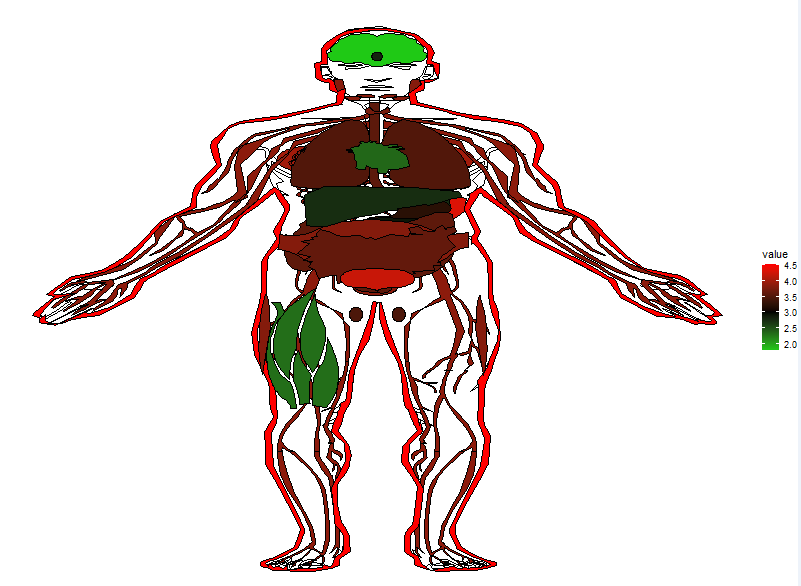
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(dplyr)) install.packages("dplyr")
if (!require(viridis)) install.packages("viridis")
if (!require(gridExtra)) install.packages("gridExtra")
if (!require(devtools)) install.packages("devtools")
devtools::install_local("gganatogram-master.zip")
# 加载库
library(gganatogram)
library(dplyr)
library(viridis)
library(gridExtra)
inputFile="male.txt"
outFile="gganatogram.tiff"
gender="male"
rt=read.table(inputFile, header=T, sep="\t", check.names=F, stringsAsFactors=F) #??ȡ?????ļ?
tiff(outFile,width=800,height=600,units="px")
gganatogram(data=rt, fillOutline='white', organism='human', sex=gender, fill="value")+ theme_void() +
scale_fill_gradient2(low = "green", mid="black", high = "red",midpoint = 3)
dev.off()效果
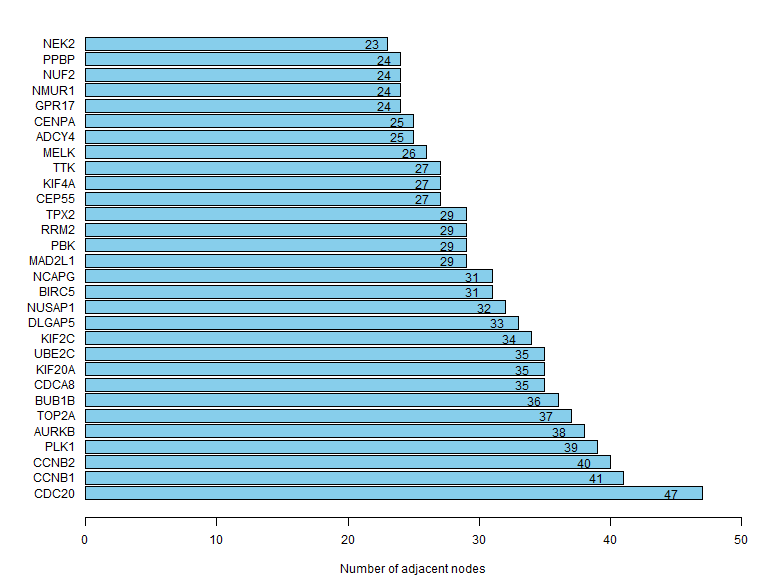
代码
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 读取输入文件
rt=read.table("input.txt", header=T, sep="\t",comment.char = "", check.names =FALSE)
tb=table(c(as.vector(rt[,1]),as.vector(rt[,2]))) #对两列进行统计
#tb=table(as.vector(rt[,1])) #对一列进行统计//不用的代码加#
tb=sort(tb,decreasing =T) #频率高的排前面
# 输入每个基因的邻接点节点数目
outTab=as.data.frame(tb)
colnames(outTab)=c("Gene","Count")
write.table(outTab,file="statResult.xls",sep="\t",quote=F,row.names=F)
# 定义柱状图显示基因数目
showNum=30
if(nrow(tb)<showNum){
showNum=nrow(outTab)
}
n=as.matrix(tb)[1:showNum,]
# 绘制柱状图
tiff(file="barplot.tiff", width = 800, height = 600, units = "px")
par(mar=c(5,7,2,3),xpd=T)
bar=barplot(n,horiz=TRUE,col="skyblue",names=FALSE,xlim=c(0,ceiling(max(n)/5)*5),xlab="Number of adjacent nodes")
text(x=n*0.95,y=bar,n)
text(x=-0.2,y=bar,label=names(n),xpd=T,pos=2)
dev.off()效果:
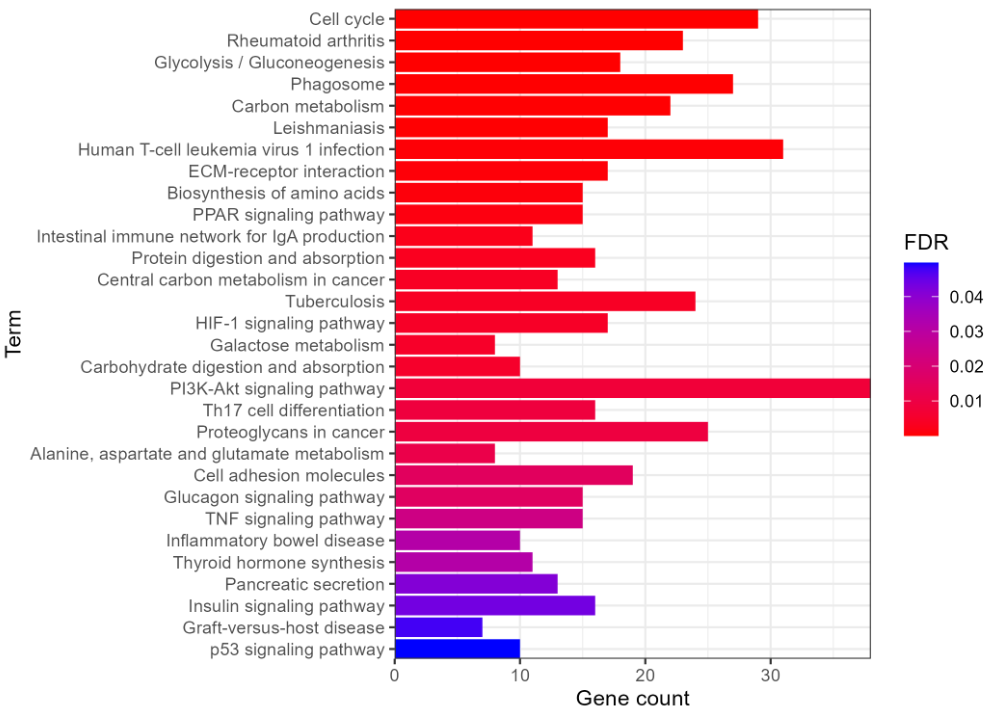
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
#三列信息需要的
#Term: GO或者通路名称
#Count: 富集在每个term的基因数目
#FDR: 矫正后的pֵ
# 加载库
library(ggplot2) #引用包
inputFile="input.txt" #输入
outFile="barplot.tiff" #输出
rt = read.table(inputFile, header=T, sep="\t", check.names=F) #读取
# 按FDR排序
labels=rt[order(rt$FDR,decreasing =T),"Term"]
rt$Term = factor(rt$Term,levels=labels)
# 绘制
p=ggplot(data=rt)+geom_bar(aes(x=Term, y=Count, fill=FDR), stat='identity')+
coord_flip() + scale_fill_gradient(low="red", high = "blue")+
xlab("Term") + ylab("Gene count") +
theme(axis.text.x=element_text(color="black", size=10),axis.text.y=element_text(color="black", size=10)) +
scale_y_continuous(expand=c(0, 0)) + scale_x_discrete(expand=c(0,0))+
theme_bw()
ggsave(outFile,width=7,height=5) #保存图片
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
inputFile="input.txt" #输入
outFile="barplot.tiff" #输出
#读取文件,整理
data=read.table(inputFile,sep="\t",header=T,row.names=1,check.names=F)
data=t(data)
col=rainbow(nrow(data),s=0.7,v=0.7)
#绘制
tiff(outFile,height=1000,width=2000, units = "px")
par(las=1,mar=c(8,5,4,16),mgp=c(3,0.1,0),cex.axis=1.5)
a1=barplot(data,col=col,yaxt="n",ylab="Relative Percent",xaxt="n",cex.lab=1.8)
a2=axis(2,tick=F,labels=F)
axis(2,a2,paste0(a2*100,"%"))
axis(1,a1,labels=F)
par(srt=60,xpd=T);text(a1,-0.02,colnames(data),adj=1,cex=0.6);par(srt=0)
ytick2 = cumsum(data[,ncol(data)])
ytick1 = c(0,ytick2[-length(ytick2)])
legend(par('usr')[2]*0.98,par('usr')[4],legend=rownames(data),col=col,pch=15,bty="n",cex=1.3)
dev.off()效果:
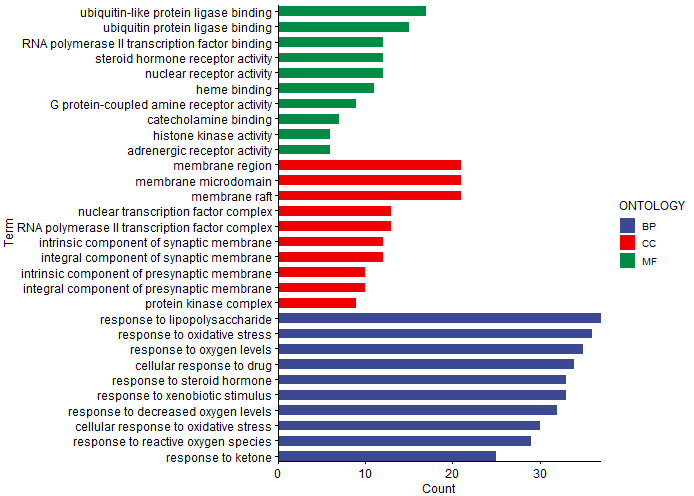
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
# 输入文件需要有3列信息
# ONTOLOGY: GO分类,BP/CC/MF
# Term: GO名称
# Count: 富集在每个Term上基因数目
# 加载库
library(ggpubr) #引用包
inputFile="input.txt" #输入
outFile="barplot.tiff" #输出
rt=read.table(inputFile,header=T,sep="\t",check.names=F) #读取输入文件
# 绘制
tiff(file=outFile,width=700,height=500, units = "px")
ggbarplot(rt, x="Term", y="Count", fill = "ONTOLOGY", color = "white",
orientation = "horiz", #横向显示
palette = "aaas", #配色方案
legend = "right", #图例位置
sort.val = "asc", #上升排序,区别于desc
sort.by.groups=TRUE)+ #按组排序
scale_y_continuous(expand=c(0, 0)) + scale_x_discrete(expand=c(0,0))
dev.off()效果:
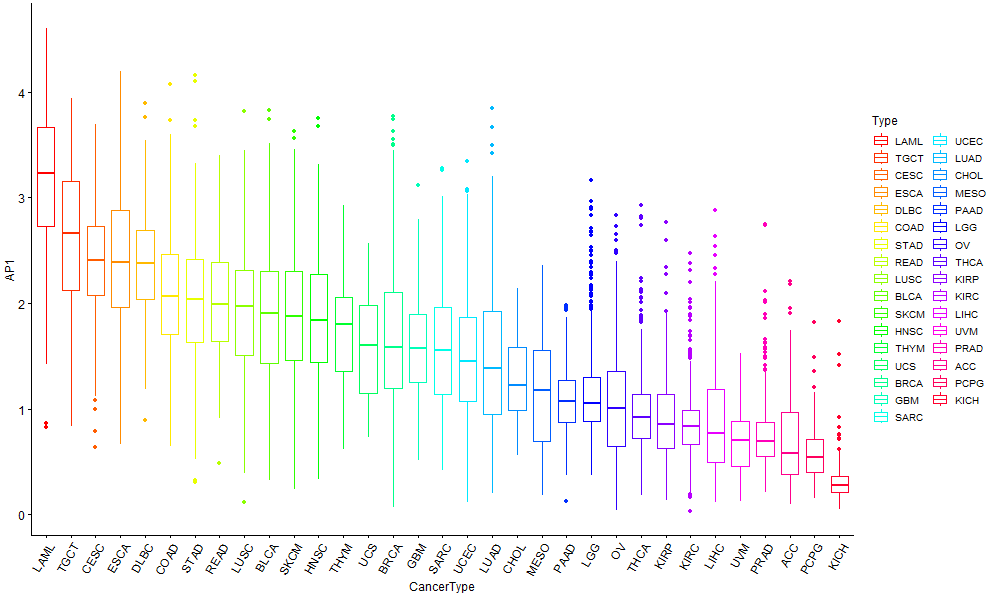
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(plyr)) install.packages("plyr")
if (!require(ggpubr)) install.packages("ggpubr")
#引用包
library(plyr)
library(ggpubr)
inputFile="input.txt" #输入
outFile="boxplot.tiff" #输出
rt=read.table(inputFile, sep="\t", header=T, check.names=F) #读取输入文件
x=colnames(rt)[2]
y=colnames(rt)[3]
colnames(rt)=c("id","Type","expression")
# 定义输出图片的排序方式
med=ddply(rt,"Type",summarise,med=median(expression))
rt$Type=factor(rt$Type, levels=med[order(med[,"med"],decreasing = T),"Type"])
# 绘制
col=rainbow(length(levels(factor(rt$Type))))
p=ggboxplot(rt, x="Type", y="expression", color = "Type",
palette = col,
ylab=y,
xlab=x,
#add = "jitter", #绘制每个样品的散点
legend = "right")
tiff(file=outFile, width=1000, height=600, units = "px") #输出图片文件
p+rotate_x_text(60) #倾斜角度
dev.off()效果:
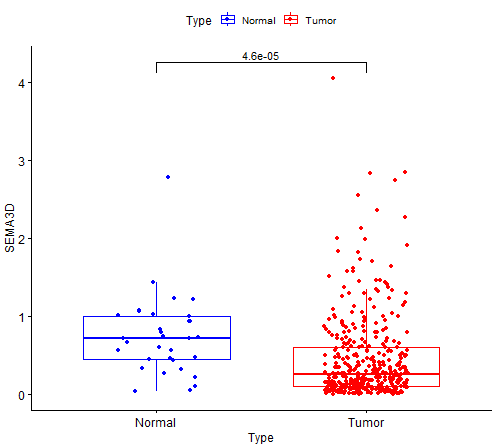
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(ggpubr) #引用包
inputFile="input.txt" #输入文件
outFile="boxplot.tiff" #输出文件
# 读取输入文件,并对输入文件整理
rt=read.table(inputFile,sep="\t",header=T,check.names=F)
x=colnames(rt)[2]
y=colnames(rt)[3]
colnames(rt)=c("id","Type","Expression")
# 设置比较租
group=levels(factor(rt$Type))
rt$Type=factor(rt$Type, levels=group)
comp=combn(group,2)
my_comparisons=list()
for(i in 1:ncol(comp)){my_comparisons[[i]]<-comp[,i]}
# 绘制boxplot
boxplot=ggboxplot(rt, x="Type", y="Expression", color="Type",
xlab=x,
ylab=y,
legend.title=x,
palette = c("blue","red"),
add = "jitter")+
stat_compare_means(comparisons = my_comparisons)
#输出图片
tiff(file=outFile,width=500,height=450, units= "px")
print(boxplot)
dev.off()效果:
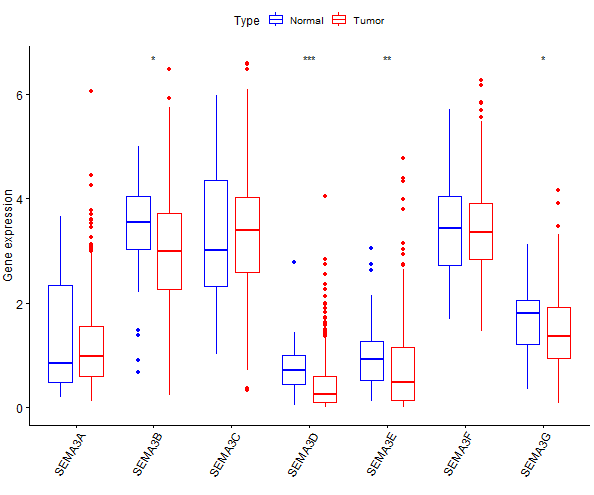
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(reshape2)) install.packages("reshape2")
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(reshape2)
library(ggpubr)
inputFile="input.txt" #输入文件
outFile="boxplot.tiff" #输出文件
#读取输入文件
rt=read.table(inputFile,sep="\t",header=T,check.names=F,row.names=1)
x=colnames(rt)[1]
colnames(rt)[1]="Type"
#把数据转换成gglpot2输入文件
data=melt(rt,id.vars=c("Type"))
colnames(data)=c("Type","Gene","Expression")
#绘制boxplot
p=ggboxplot(data, x="Gene", y="Expression", color = "Type",
ylab="Gene expression",
xlab="",
legend.title=x,
palette = c("blue","red"),
width=0.6, add = "none")
p=p+rotate_x_text(60)
p1=p+stat_compare_means(aes(group=Type),
method="wilcox.test",
symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", " ")),
label = "p.signif")
#输出图片
tiff(file=outFile, width=600, height=500, units = "px")
print(p1)
dev.off()效果:
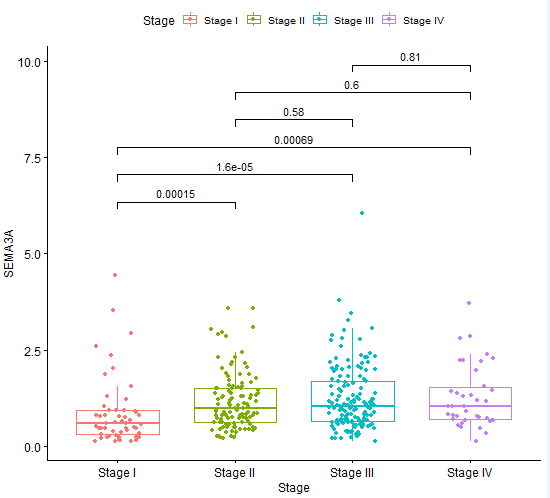
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(ggpubr)
inputFile="input.txt"
outFile="boxplot.tiff"
#读取输入文件
rt=read.table(inputFile,sep="\t",header=T,check.names=F)
x=colnames(rt)[2]
y=colnames(rt)[3]
colnames(rt)=c("id","Type","Expression")
#设置比较租
group=levels(factor(rt$Type))
rt$Type=factor(rt$Type, levels=group)
comp=combn(group,2)
my_comparisons=list()
for(i in 1:ncol(comp)){my_comparisons[[i]]<-comp[,i]}
#绘制boxplot
boxplot=ggboxplot(rt, x="Type", y="Expression", color="Type",
xlab=x,
ylab=y,
legend.title=x,
add = "jitter")+
stat_compare_means(comparisons = my_comparisons)
#输出图片
tiff(file=outFile, width=550, height=500, units = "px")
print(boxplot)
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(reshape2)) install.packages("reshape2")
if (!require(ggplot2)) install.packages("ggplot2")
# 加载库
library(ggplot2)
library(reshape2)
inputFile="input.txt"
outFile="boxplot.tiff"
#读取输入文件
rt=read.table(inputFile, header=T,sep="\t",check.names=F,row.names=1)
x=colnames(rt)[1]
colnames(rt)[1]="Type"
#差异分析
geneSig=c("")
for(gene in colnames(rt)[2:ncol(rt)]){
rt1=rt[,c(gene,"Type")]
colnames(rt1)=c("expression","Type")
p=1
if(length(levels(factor(rt1$Type)))>2){
test=kruskal.test(expression ~ Type, data = rt1)
p=test$p.value
}else{
test=wilcox.test(expression ~ Type, data = rt1)
p=test$p.value
}
Sig=ifelse(p<0.001,"***",ifelse(p<0.01,"**",ifelse(p<0.05,"*","")))
geneSig=c(geneSig,Sig)
}
colnames(rt)=paste0(colnames(rt),geneSig)
#把数据转换成ggplot2输入文件
data=melt(rt,id.vars=c("Type"))
colnames(data)=c("Type","Gene","Expression")
#绘制
p1=ggplot(data,aes(x=Type,y=Expression,fill=Type))+
guides(fill=guide_legend(title=x))+
labs(x = x, y = "Gene expression")+
geom_boxplot()+ facet_wrap(~Gene,nrow =1)+ theme_bw()+
theme(axis.text.x = element_text(angle = 45, hjust = 1))
#输出
tiff(file=outFile, width=900, height=500, units = "px")
print(p1)
dev.off()效果:
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(ggpubr)
inputFile="input.txt"
outFile="vioplot.tiff"
#读取
rt=read.table(inputFile,header=T,sep="\t",check.names=F)
x=colnames(rt)[2]
y=colnames(rt)[3]
colnames(rt)=c("id","Type","Expression")
#s设置比较组
group=levels(factor(rt$Type))
rt$Type=factor(rt$Type, levels=group)
comp=combn(group,2)
my_comparisons=list()
for(i in 1:ncol(comp)){my_comparisons[[i]]<-comp[,i]}
#绘制
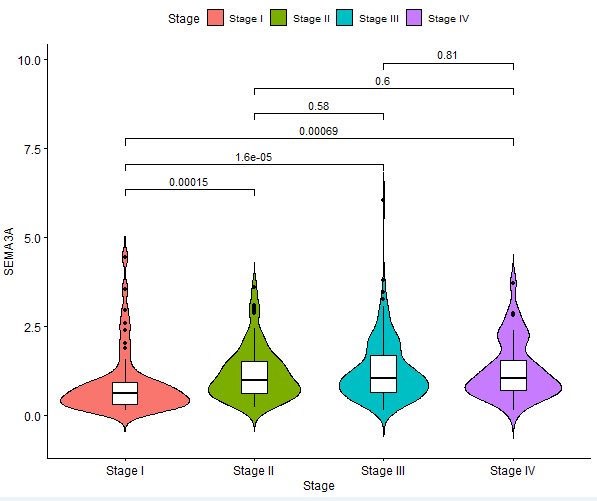
tiff(file=outFile, width=600, height=500, units = "px")
ggviolin(rt, x="Type", y="Expression", fill = "Type",
xlab=x, ylab=y,
legend.title=x,
add = "boxplot", add.params = list(fill="white"))+
stat_compare_means(comparisons = my_comparisons)
#stat_compare_means(comparisons = my_comparisons,symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", "ns")),label = "p.signif")
dev.off()效果:
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(reshape2)) install.packages("reshape2")
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(reshape2)
library(ggpubr)
inputFile="input.txt"
outFile="vioplot.tiff"
#读取文件
rt=read.table(inputFile,header=T,sep="\t",check.names=F,row.names=1)
x=colnames(rt)[1]
colnames(rt)[1]="Type"
#把数据转换成ggplot2数据文件
data=melt(rt,id.vars=c("Type"))
colnames(data)=c("Type","Gene","Expression")
#绘制小提琴图
p=ggviolin(data, x="Gene", y="Expression", color = "Type",
ylab="Gene expression",
xlab=x,
legend.title=x,
add.params = list(fill="white"),
palette = c("blue","red"),
width=1, add = "boxplot")
p=p+rotate_x_text(60)
p1=p+stat_compare_means(aes(group=Type),
method="wilcox.test",
symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", " ")),
label = "p.signif")
#输出
tiff(file=outFile, width=600, height=500, units = "px")
print(p1)
dev.off()效果:

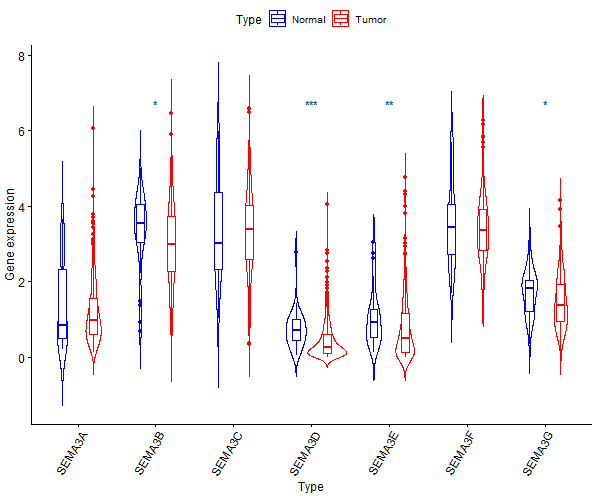
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(reshape2)) install.packages("reshape2")
if (!require(ggplot2)) install.packages("ggplot2")
# 加载库
library(ggplot2)
library(reshape2)
inputFile="input.txt"
outFile="vioplot.tiff"
#读取输入文件
rt=read.table(inputFile, header=T,sep="\t",check.names=F,row.names=1)
x=colnames(rt)[1]
colnames(rt)[1]="Type"
#差异分析
geneSig=c("")
for(gene in colnames(rt)[2:ncol(rt)]){
rt1=rt[,c(gene,"Type")]
colnames(rt1)=c("expression","Type")
p=1
if(length(levels(factor(rt1$Type)))>2){
test=kruskal.test(expression ~ Type, data = rt1)
p=test$p.value
}else{
test=wilcox.test(expression ~ Type, data = rt1)
p=test$p.value
}
Sig=ifelse(p<0.001,"***",ifelse(p<0.01,"**",ifelse(p<0.05,"*","")))
geneSig=c(geneSig,Sig)
}
colnames(rt)=paste0(colnames(rt),geneSig)
# 把数据转换成ggplot2文件
data=melt(rt,id.vars=c("Type"))
colnames(data)=c("Type","Gene","Expression")
# 绘制
p1=ggplot(data,aes(x=Type,y=Expression,fill=Type))+
guides(fill=guide_legend(title=x))+
labs(x = x, y = "Gene expression")+
geom_violin()+ geom_boxplot(width=0.2,position=position_dodge(0.9))+
facet_wrap(~Gene,nrow =1)+ theme_bw()+
theme(axis.text.x = element_text(angle = 45, hjust = 1))
# 出图
tiff(file=outFile, width=900, height=500, units = "px")
print(p1)
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library("ggpubr")
inputFile="input.txt"
outFile="pairDiff.tiff"
ylab="BRCA1 expression" #y轴名称
#读取输入文件
data=read.table(inputFile,sep="\t",header=T,check.names=F,row.names=1)
cond1=colnames(data)[1]
cond2=colnames(data)[2]
#绘制
tiff(file=outFile, width=500, height=400, units = "px")
ggpaired(data, cond1 = cond1, cond2 = cond2, fill = "condition", palette = "jco",
legend.title="Condition",xlab="",ylab = ylab)+
#stat_compare_means(paired = TRUE, label = "p.format", label.x = 1.35)
stat_compare_means(paired = TRUE, symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1), symbols = c("***", "**", "*", "ns")),label = "p.signif",label.x = 1.35)
dev.off()效果:
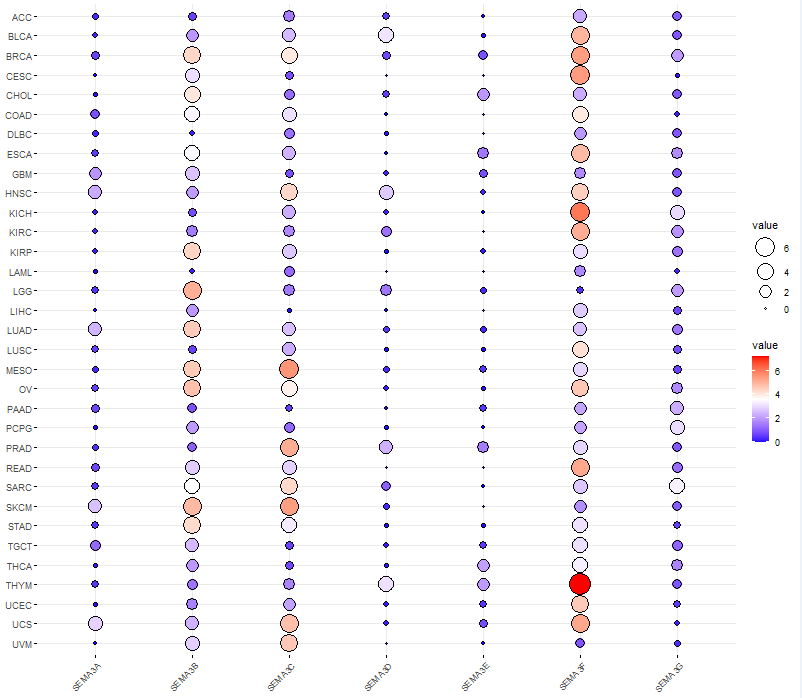
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(ggpubr)
inputFile="input.txt"
outFile="ggballoonplot.tiff" #输出文件
data=read.table(inputFile,sep="\t",header=T,check.names=F,row.names=1)
# 绘制气泡图
tiff(file=outFile,width=800,height=700, units = "px")
ggballoonplot(data, fill = 'value')+gradient_fill(c('blue', 'white', 'red'))
dev.off()效果:
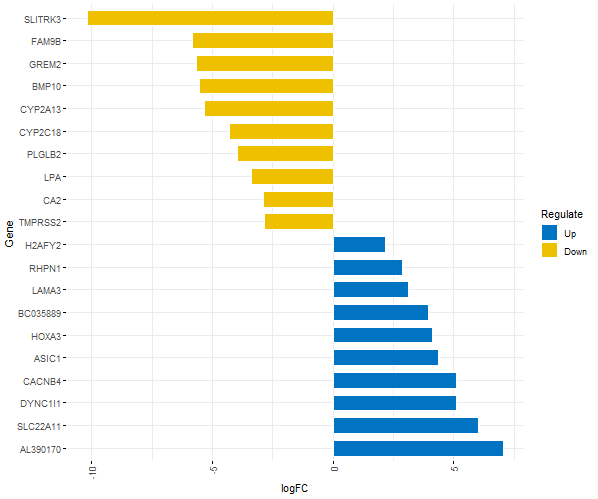
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
# 加载库
library(ggpubr)
inputFile="input.txt"
outFile="Deviation.tiff"
# 读取
rt=read.table(inputFile,sep="\t",header=T,check.names=F)
x=colnames(rt)[1]
y=colnames(rt)[2]
colnames(rt)=c("Name","Value")
rt$Regulate=factor(ifelse(rt$Value<0, "Down", "Up"), levels = c("Up", "Down"))
# 绘制
tiff(file=outFile,width=600,height=500, units = "px")
ggbarplot(rt, x="Name", y="Value", fill = "Regulate", color = "white",
palette = "jco",
sort.val = "desc", sort.by.groups = FALSE,
x.text.angle=90,
xlab = x, ylab = y,
legend.title="Regulate", rotate=TRUE, ggtheme = theme_minimal()) #rotatex设置x/y对调
dev.off()效果:
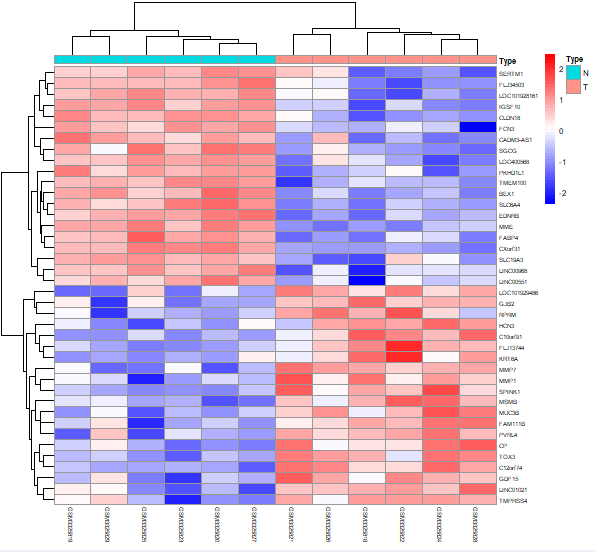
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(pheatmap)) install.packages("pheatmap")
# 加载库
library(pheatmap)
inputFile="input.txt"
groupFile="group.txt" #分组文件
outFile="heatmap.tiff"
rt=read.table(inputFile,header=T,sep="\t",row.names=1,check.names=F) #读取文件
ann=read.table(groupFile,header=T,sep="\t",row.names=1,check.names=F) #读取样本属性文件
# 绘制
tiff(file=outFile,width=600,height=550, units = "px")
pheatmap(rt,
annotation=ann,
cluster_cols = T,
color = colorRampPalette(c("blue", "white", "red"))(50),
show_colnames = T,
scale="row", #矫正
#border_color ="NA",
fontsize = 8,
fontsize_row=6,
fontsize_col=6)
dev.off()效果:
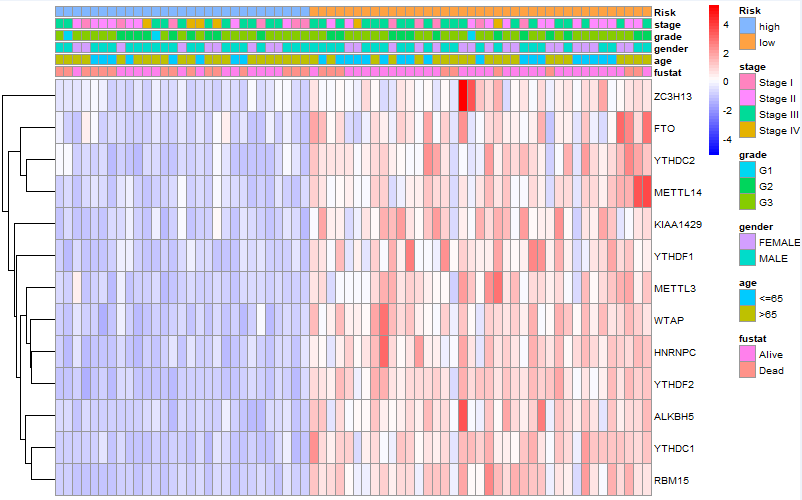
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(pheatmap)) install.packages("pheatmap")
# 加载库
library(pheatmap)
expFile="input.txt"
cliFile="clinical.txt"
outFile="heatmap.tiff"
var="Risk" #按照临床性状(此处为风险)对样品排序
rt=read.table(expFile, sep="\t", header=T, row.names=1, check.names=F) #读取表达文件
Type=read.table(cliFile, sep="\t", header=T, row.names=1, check.names=F) #读取临床文件
#样品取交集
sameSample=intersect(colnames(rt),row.names(Type))
rt=rt[,sameSample]
Type=Type[sameSample,]
Type=Type[order(Type[,var]),] #按临床性状排序
rt=rt[,row.names(Type)]
#绘制热图
tiff(outFile,height=500,width=800, units = "px")
pheatmap(rt, annotation=Type,
color = colorRampPalette(c("blue", "white", "red"))(50),
cluster_cols =F, #是否聚类
scale="row", #基因矫正
show_colnames=F,
fontsize=10,
fontsize_row=10,
fontsize_col=8)
dev.off()效果:
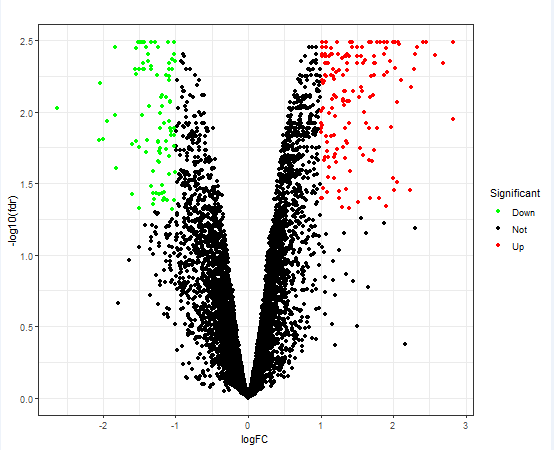
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
# 加载库
library(ggplot2)
inputFile="input.txt"
outFile="vol.tiff"
logFCfilter=1 #logFC过滤阈值
fdrFilter=0.05 #矫正后p值阈值ֵ
#读取文件
rt=read.table(inputFile,sep="\t",header=T,check.names=F)
#定义显著性
Significant=ifelse((rt$fdr<fdrFilter & abs(rt$logFC)>logFCfilter), ifelse(rt$logFC>logFCfilter,"Up","Down"), "Not")
#绘制火山图
p = ggplot(rt, aes(logFC, -log10(fdr)))+
geom_point(aes(col=Significant))+
scale_color_manual(values=c("green", "black", "red"))+
labs(title = " ")+
theme(plot.title = element_text(size = 16, hjust = 0.5, face = "bold"))
p=p+theme_bw()
#出图
tiff(outFile,width=550,height=450)
print(p)
dev.off()效果:
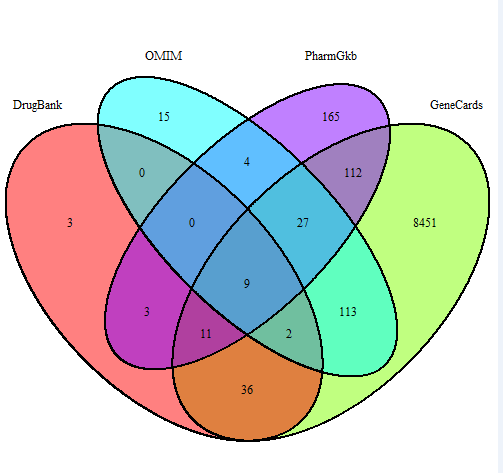
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(VennDiagram)) install.packages("VennDiagram")
# 加载库
library(VennDiagram) #引用包
outFile="intersectGenes.txt" #输出交集基因文件
outPic="venn.tiff" #输出图片文件
files=dir() #获取目录下所有文件
files=grep("txt$",files,value=T) #提取TXT结尾的文件
geneList=list()
# 读取所有txt文件中的基因信息,保存到GENELIST
for(i in 1:length(files)){
inputFile=files[i]
if(inputFile==outFile){next}
rt=read.table(inputFile,header=F) #读取
geneNames=as.vector(rt[,1]) #提取基因名
geneNames=gsub("^ | $","",geneNames) #去掉基因首尾的空格
uniqGene=unique(geneNames) #基因取unique
header=unlist(strsplit(inputFile,"\\.|\\-"))
geneList[[header[1]]]=uniqGene
uniqLength=length(uniqGene)
print(paste(header[1],uniqLength,sep=" "))
}
# 绘制vennͼ
venn.plot=venn.diagram(geneList,filename=NULL,fill=rainbow(length(geneList)) )
tiff(file=outPic, width=500, height=500)
grid.draw(venn.plot)
dev.off()
# 保存交集基因
intersectGenes=Reduce(intersect,geneList)
write.table(file=outFile,intersectGenes,sep="\t",quote=F,col.names=F,row.names=F)效果:
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(UpSetR)) install.packages("UpSetR")
# 加载库
library(UpSetR)
outFile="intersectGenes.txt" #输出交集基因文件
outPic="upset.tiff" #输出图片
files=dir() #获取目录下所有文件
files=grep("txt$",files,value=T) #提取.txt结尾的文件
geneList=list()
#获取所有txt文件中的基因信息,保存到geneList
for(i in 1:length(files)){
inputFile=files[i]
if(inputFile==outFile){next}
rt=read.table(inputFile,header=F) #读取输入文件
geneNames=as.vector(rt[,1]) #提取基因名称
geneNames=gsub("^ | $","",geneNames) #去掉基因首尾的空格
uniqGene=unique(geneNames) #基因取unique,唯一基因列表
header=unlist(strsplit(inputFile,"\\.|\\-"))
geneList[[header[1]]]=uniqGene
uniqLength=length(uniqGene)
print(paste(header[1],uniqLength,sep=" "))
}
#绘制UpSetͼ
upsetData=fromList(geneList)
tiff(file=outPic,width=900,height=600)
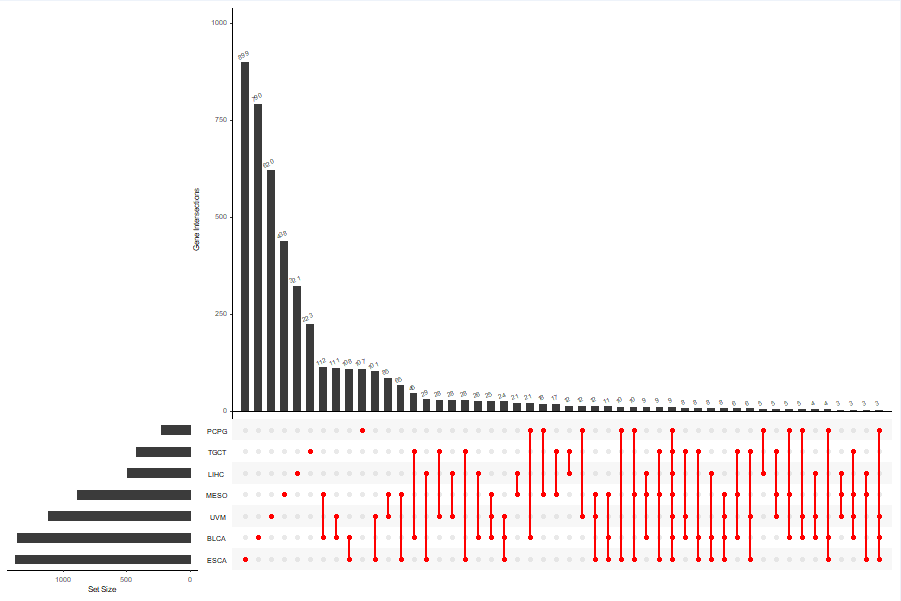
upset(upsetData,
nsets = length(geneList), #展示多少个数据
nintersects = 50, #展示基因集数目
order.by = "freq", #按照数目排序
show.numbers = "yes", #柱状图上方是否显示数值
number.angles = 20, #字体角度
point.size = 2, #点的大小
matrix.color="red", #交集点颜色
line.size = 0.8, #线条粗细
mainbar.y.label = "Gene Intersections",
sets.x.label = "Set Size")
dev.off()
#保存交集文件
intersectGenes=Reduce(intersect,geneList)
write.table(file=outFile,intersectGenes,sep="\t",quote=F,col.names=F,row.names=F)效果:
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(ggpubr)) install.packages("ggpubr")
if (!require(ggExtra)) install.packages("ggExtra")
# 加载库
library(ggplot2)
library(ggpubr)
library(ggExtra)
inputFile="input.txt"
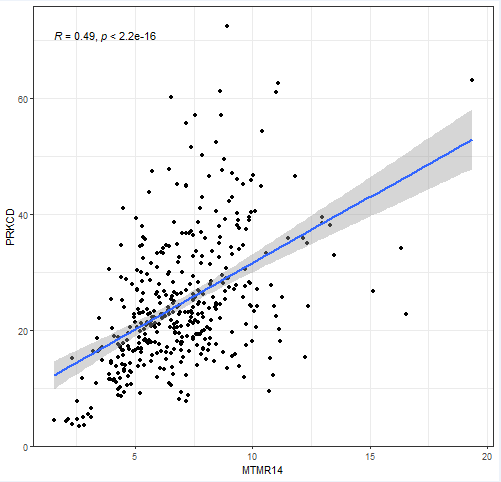
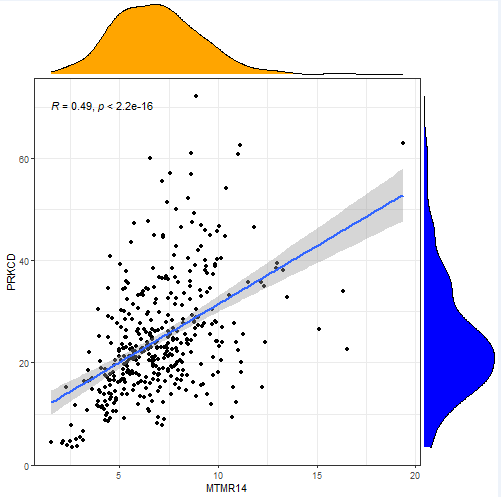
gene1="MTMR14" #第一个基因名字
gene2="PRKCD" #第二个基因名字
#读取输入文件,提取基因表达量
rt=read.table(inputFile,sep="\t",header=T,check.names=F,row.names=1)
x=as.numeric(rt[gene1,])
y=as.numeric(rt[gene2,])
#相关性分析
df1=as.data.frame(cbind(x,y))
corT=cor.test(x,y,method="spearman")
cor=corT$estimate
pValue=corT$p.value
p1=ggplot(df1, aes(x, y)) +
xlab(gene1)+ylab(gene2)+
geom_point()+ geom_smooth(method="lm",formula = y ~ x) + theme_bw()+
stat_cor(method = 'spearman', aes(x =x, y =y))
p2=ggMarginal(p1, type = "density", xparams = list(fill = "orange"),yparams = list(fill = "blue"))
#出图
tiff(file="cor.tiff",width=500,height=480)
print(p1)
dev.off()
#出图2
tiff(file="cor.density.tiff",width=500,height=500)
print(p2)
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(corrplot)) install.packages("corrplot")
# 加载库
library(corrplot)
inputFile="input.txt"
rt=read.table(inputFile,sep="\t",header=T,row.names=1) #读取文件
rt=t(rt) #数据转置
M=cor(rt) #相关型矩阵
#绘制相关性图形
tiff(file="corpot1.tiff",width=700,height=700)
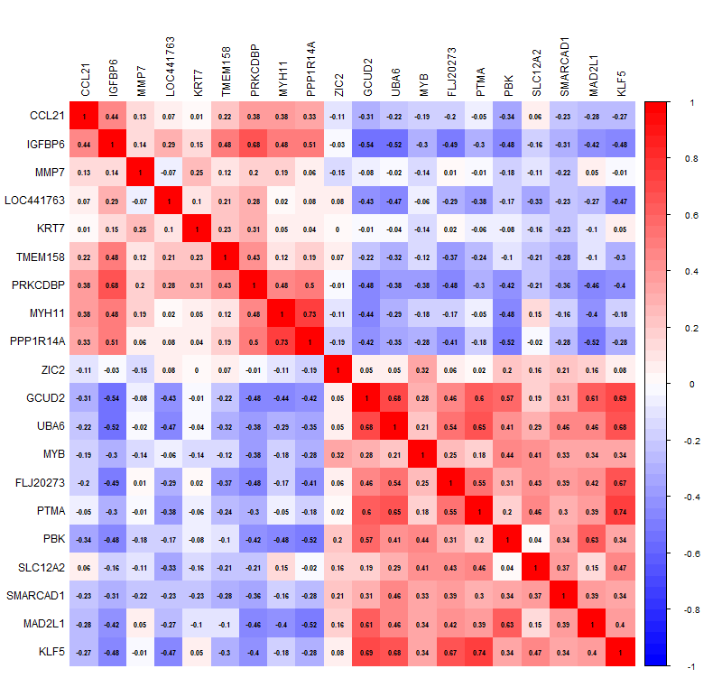
corrplot(M,
method = "circle",
order = "hclust", #聚类
type = "upper",
col=colorRampPalette(c("green", "white", "red"))(50)
)
dev.off()
#第二个图
tiff(file="corpot2.tiff",width=800,height=800)
corrplot(M,
order="original",
method = "color",
number.cex = 0.7, #相关系数
addCoef.col = "black",
diag = TRUE,
tl.col="black",
col=colorRampPalette(c("blue", "white", "red"))(50))
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(corrplot)) install.packages("corrplot")
if (!require(circlize)) install.packages("circlize")
# 加载库
options(stringsAsFactors=F)
library(corrplot)
library(circlize)
inputFile="input.txt"
outFile="circos.tiff"
#读取输入文件
rt=read.table(inputFile,sep="\t",header=T,check.names=F,row.names=1)
rt=t(rt) #转置
#计算基因间相关系数
cor1=cor(rt)
#设置图形颜色
col = c(rgb(1,0,0,seq(1,0,length=32)),rgb(0,1,0,seq(0,1,length=32)))
cor1[cor1==1]=0 #删掉相关性=1
c1 = ifelse(c(cor1)>=0,rgb(1,0,0,abs(cor1)),rgb(0,1,0,abs(cor1)))
col1 = matrix(c1,nc=ncol(rt))
#绘制圈图
tiff(outFile,width=700,height=700)
par(mar=c(2,2,2,4))
circos.par(gap.degree=c(3,rep(2, nrow(cor1)-1)), start.degree = 180)
chordDiagram(cor1, grid.col=rainbow(ncol(rt)), col=col1, transparency = 0.5, symmetric = T)
par(xpd=T)
colorlegend(col, vertical = T,labels=c(1,0,-1),xlim=c(1.1,1.3),ylim=c(-0.4,0.4)) #????ͼ??
dev.off()
circos.clear()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(igraph)) install.packages("igraph")
if (!require(reshape2)) install.packages("reshape2")
# 加载库
library(igraph)
library(reshape2)
inputFile="input.txt"
outFile="corNetwork.tiff"
cutoff=0.4 #相关性阈值
#读取输入文件
data=read.table(inputFile,header=T,sep="\t",row.names=1,check.names=F)
cordata=cor(t(data))
#保留相关性矩阵的一半
mydata = cordata
upper = upper.tri(mydata)
mydata[upper] = NA
#把相关性矩阵转换为数据框
df = data.frame(gene=rownames(mydata),mydata)
dfmeltdata = melt(df,id="gene")
dfmeltdata = dfmeltdata[!is.na(dfmeltdata$value),]
dfmeltdata = dfmeltdata[dfmeltdata$gene!=dfmeltdata$variable,]
dfmeltdata = dfmeltdata[abs(dfmeltdata$value)>cutoff,]
#定义网络图的节点和边
corweight = dfmeltdata$value
weight = corweight+abs(min(corweight))+5
d = data.frame(p1=dfmeltdata$gene,p2=dfmeltdata$variable,weight=dfmeltdata$value)
g = graph.data.frame(dfmeltdata,directed = FALSE)
#设置颜色,节点大小,字体大小
E(g)$color = ifelse(corweight>0,rgb(254/255,67/255,101/255,abs(corweight)),rgb(0/255,0/255,255/255,abs(corweight)))
V(g)$size = 8
V(g)$shape = "circle"
V(g)$lable.cex = 1.2
V(g)$color = "white"
E(g)$weight = weight
#可视化
tiff(outFile,width=700,height=600)
layout(matrix(c(1,1,1,0,2,0),byrow=T,nc=3),height=c(6,1),width=c(3,4,3))
par(mar=c(1.5,2,2,2))
vertex.frame.color = NA
plot(g,layout=layout_nicely,vertex.label.cex=V(g)$lable.cex,edge.width = E(g)$weight,edge.arrow.size=0,vertex.label.color="black",vertex.frame.color=vertex.frame.color,edge.color=E(g)$color,vertex.label.cex=V(g)$lable.cex,vertex.label.font=2,vertex.size=V(g)$size,edge.curved=0.4)
#绘制图例
color_legend = c(rgb(254/255,67/255,101/255,seq(1,0,by=-0.01)),rgb(0/255,0/255,255/255,seq(0,1,by=0.01)))
par(mar=c(2,2,1,2),xpd = T,cex.axis=1.6,las=1)
barplot(rep(1,length(color_legend)),border = NA, space = 0,ylab="",xlab="",xlim=c(1,length(color_legend)),horiz=FALSE,
axes = F, col=color_legend,main="")
axis(3,at=seq(1,length(color_legend),length=5),c(1,0.5,0,-0.5,-1),tick=FALSE)
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(fmsb)) install.packages("fmsb")
# 加载库
library(fmsb)
inputFile="input.txt"
outFile="radar.tiff"
col="red" #定义颜色
data=read.table(inputFile,header=T,sep="\t",row.names=1,check.names=F)
data=as.data.frame(t(data)) #转置
maxValue=ceiling(max(abs(data))*10)/10 #设置刻度
data=rbind(rep(maxValue,ncol(data)),rep(-maxValue,ncol(data)),data)
#输出结果
tiff(file=outFile,height=700,width=700)
radarchart( data, axistype=1,
pcol=col, #线条颜色
plwd=2 , #线条粗细
plty=1, #虚线,实线
cglcol="grey", #背景线条颜色
cglty=1, #背景线条虚线,实线1
caxislabels=seq(-maxValue,maxValue,maxValue/2), #坐标刻度
cglwd=1.2, #背景线条粗细
axislabcol="blue", #刻度颜色
vlcex=0.8 #字体大小
)
dev.off()效果:
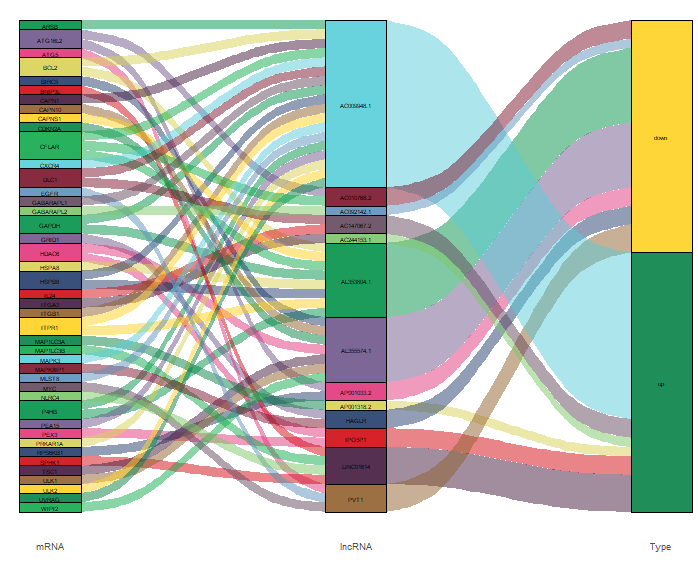
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggalluvial)) install.packages("ggalluvial")
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(dplyr)) install.packages("dplyr")
# 加载库
library(ggalluvial)
library(ggplot2)
library(dplyr)
inputFile="input.txt"
outFile="ggalluvial.tiff"
rt=read.table(inputFile, header = T, sep="\t", check.names=F)
corLodes=to_lodes_form(rt, axes = 1:ncol(rt), id = "Cohort")
#得到输出文件
tiff(file=outFile,width=700,height=600)
mycol <- rep(c("#029149","#6E568C","#E0367A","#D8D155","#223D6C","#D20A13","#431A3D","#91612D","#FFD121","#088247","#11AA4D","#58CDD9","#7A142C","#5D90BA","#64495D","#7CC767"),15)
ggplot(corLodes, aes(x = x, stratum = stratum, alluvium = Cohort,fill = stratum, label = stratum)) +
scale_x_discrete(expand = c(0, 0)) +
#用aes.flow控制线调颜色,forward说明颜色和前面一致,backward说明与后面一致
geom_flow(width = 2/10,aes.flow = "forward") +
geom_stratum(alpha = .9,width = 2/10) +
scale_fill_manual(values = mycol) +
#size = 2代表基因名字大小
geom_text(stat = "stratum", size = 2,color="black") +
xlab("") + ylab("") + theme_bw() +
theme(axis.line = element_blank(),axis.ticks = element_blank(),axis.text.y = element_blank()) + #ȥ????????
theme(panel.grid =element_blank()) +
theme(panel.border = element_blank()) +
ggtitle("") + guides(fill = FALSE)
dev.off()效果:
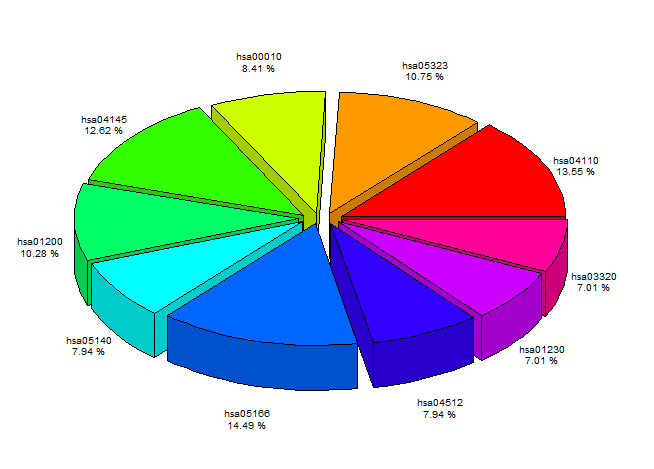
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(plotrix)) install.packages("plotrix")
# 加载库
library("plotrix")
inputFile="input.txt"
outFile="pie.tiff"
#读取文件,整理
rt=read.table(inputFile,header=T,sep="\t",check.names=F)
x=rt[,2]
labels=as.character(rt[,1])
#求个部分百分率
piepercent=paste(round(100*x/sum(x), 2), "%")
labels=paste0(labels,"\n",piepercent)
#绘制饼图
tiff(file = outFile,width=700,height=600) #保存图片
pie3D(x, labels = labels, height=0.1, labelcex=0.8,
explode =0.1, theta=0.85) #explode:离中心距离 theta:观看角度
dev.off()效果:
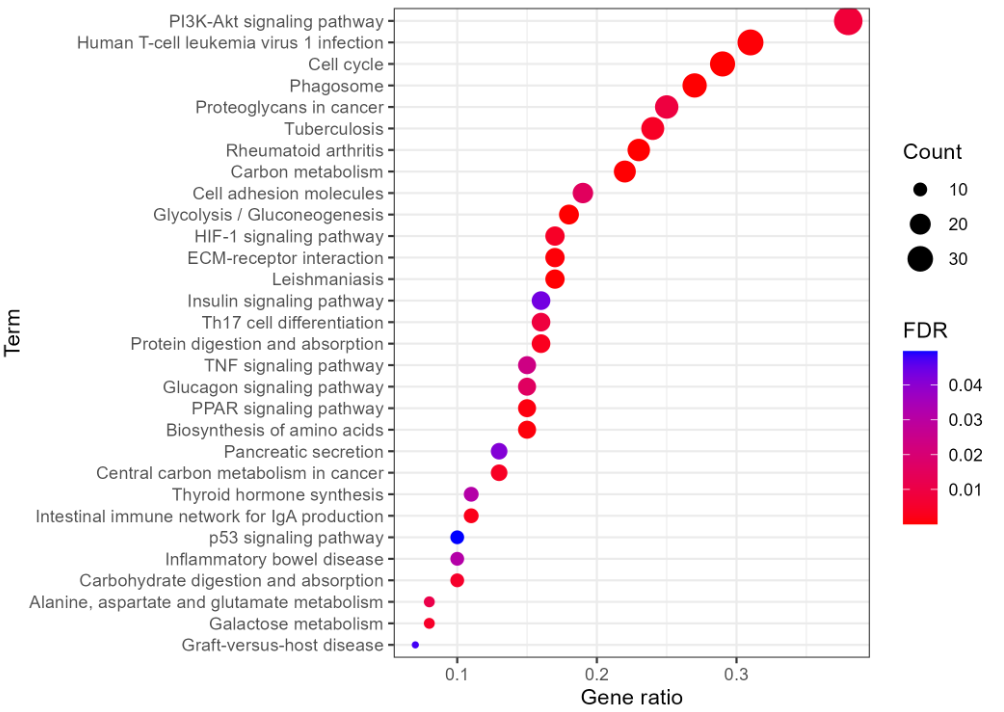
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
#输入文件需要四列信息
#Term: GO或名称
#Ratio: 基因比例
#Count: 富集数目
#FDR: 矫正pֵ
# 加载库
library(ggplot2)
inputFile="input.txt"
outFile="bubble.tiff"
rt = read.table(inputFile, header=T, sep="\t", check.names=F)
#按照Ratio对Term排序
labels=rt[order(rt$Ratio),"Term"]
rt$Term = factor(rt$Term,levels=labels)
# 绘制
p = ggplot(rt,aes(Ratio, Term)) +
geom_point(aes(size=Count, color=FDR))
p1 = p +
scale_colour_gradient(low="red",high="blue") +
labs(color="FDR",size="Count",x="Gene ratio",y="Term")+
theme(axis.text.x=element_text(color="black", size=10),axis.text.y=element_text(color="black", size=10)) +
theme_bw()
ggsave(outFile, width=7, height=5) #保存效果:
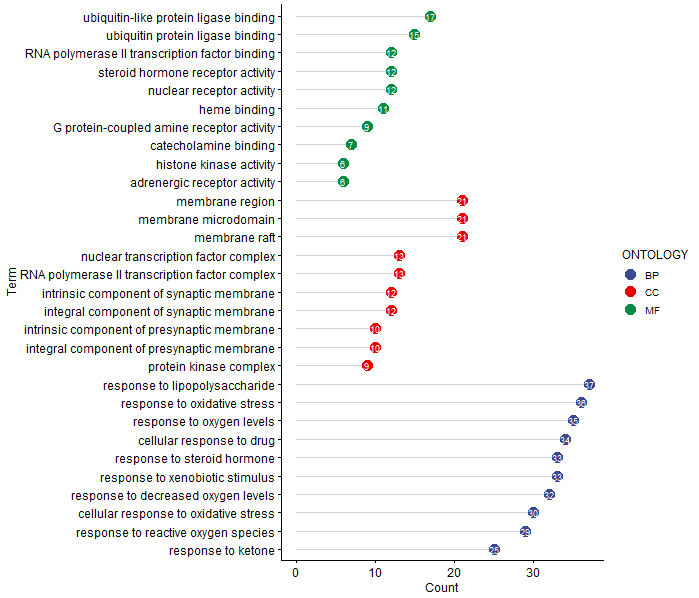
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggpubr)) install.packages("ggpubr")
#输入文件需要有三列信息
#ONTOLOGY: GO分类(BP, CC, MF)
#Term: GO名称
#Count: 富集在每个GO上的数目
library(ggpubr)
inputFile="input.txt"
outFile="Lollipop.tiff"
rt=read.table(inputFile,header=T,sep="\t",check.names=F) #读取
#绘制棒棒糖图
tiff(file=outFile,width=700,height=600)
ggdotchart(rt, x="Term", y="Count", color = "ONTOLOGY",group = "ONTOLOGY",
palette = "aaas", #配色方案
legend = "right", #图例位置
sorting = "descending", #上升排序,区别于desc
add = "segments", #增加线段
rotate = TRUE, #横向显示
dot.size = 5, #圆圈大小
label = round(rt$Count), #圆圈内数值
font.label = list(color="white",size=9, vjust=0.5), #圆圈内数值字体设置
ggtheme = theme_pubr())
dev.off()效果:
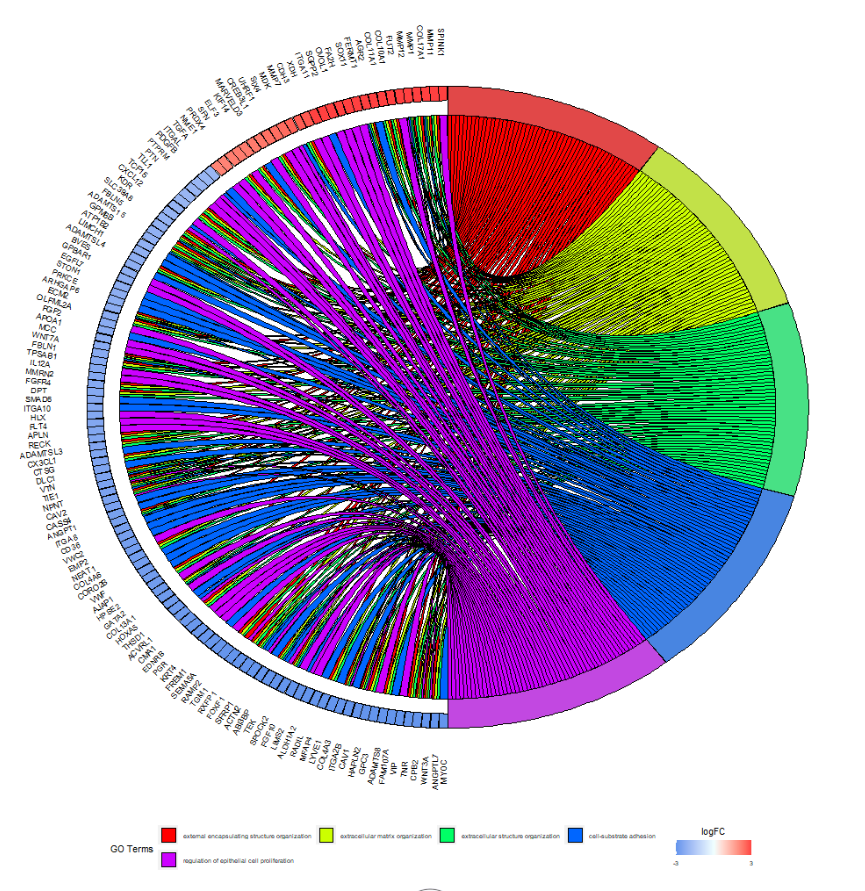
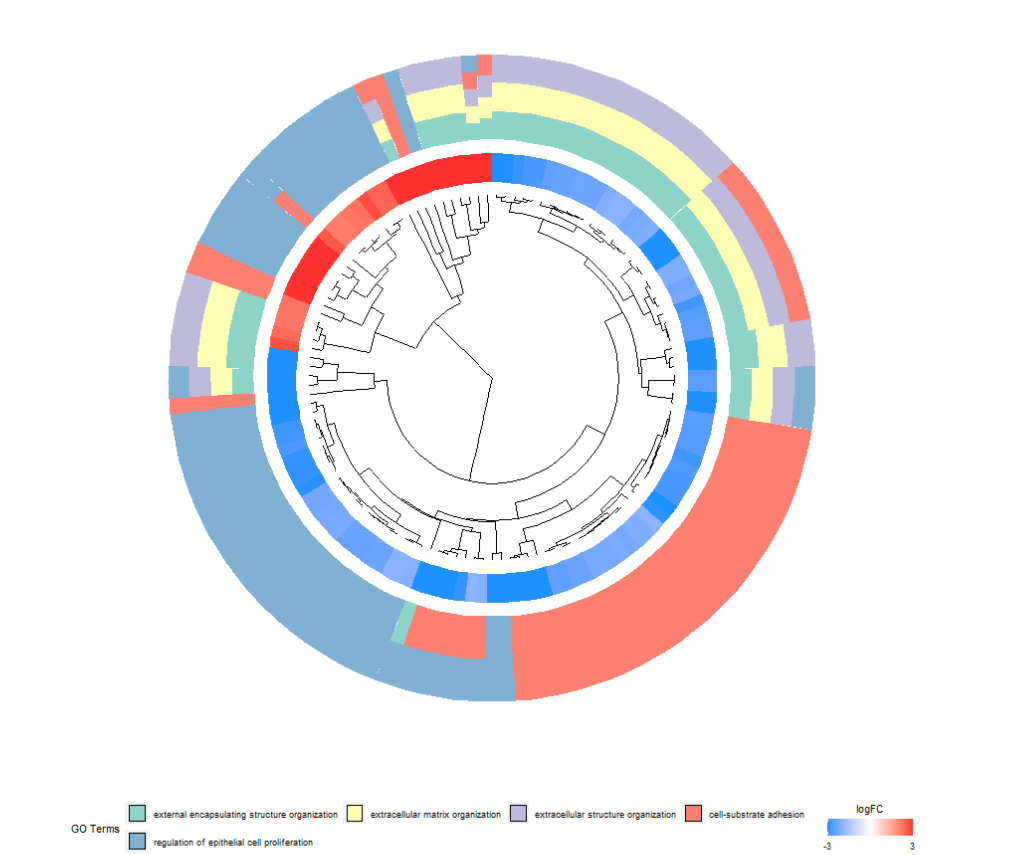
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(colorspace)) install.packages("colorspace")
if (!require(stringi)) install.packages("stringi")
if (!require(digest)) install.packages("digest")
if (!require(GOplot)) install.packages("GOplot")
if (!require(BiocManager)) install.packages("BiocManager")
# 生信的专用库
if (!require(org.Hs.eg.db)) BiocManager::install("org.Hs.eg.db")
if (!require(DOSE)) BiocManager::install("DOSE")
if (!require(clusterProfiler)) BiocManager::install("clusterProfiler")
if (!require(enrichplot)) BiocManager::install("enrichplot")
# 加载库
library(clusterProfiler)
library(org.Hs.eg.db)
library(enrichplot)
library(ggplot2)
library(GOplot)
#过滤
pFilter=0.05
adjPfilter=0.05
inputFile="input.txt"
rt=read.table(inputFile,sep="\t",header=T,check.names=F)
genes=as.vector(rt[,1])
entrezIDs=mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA) #找出基因对应的id
entrezIDs=as.character(entrezIDs)
rt=cbind(rt,entrezID=entrezIDs)
rt=rt[is.na(rt[,"entrezID"])==F,] #删掉没有基因ID的
gene=rt$entrezID
# GO
GO=enrichGO(gene = gene,
OrgDb = org.Hs.eg.db,
pvalueCutoff =1,
qvalueCutoff = 1,
ont="all",
readable =T)
GO=as.data.frame(GO)
GO=GO[(GO$pvalue<pFilter & GO$p.adjust<adjPfilter),]
write.table(GO,file="GO.txt",sep="\t",quote=F,row.names = F) #
# GO
go=data.frame(Category = GO$ONTOLOGY,ID = GO$ID,Term = GO$Description, Genes = gsub("/", ", ", GO$geneID), adj_pval = GO$p.adjust)
# logFC
genelist=data.frame(ID = rt$gene, logFC = rt$logFC)
row.names(genelist)=genelist[,1]
circ <- circle_dat(go, genelist)
termNum = 5 #限定GO数目
termNum=ifelse(nrow(go)<termNum,nrow(go),termNum)
geneNum = nrow(genelist) #限定基因数目
# 绘制
chord <- chord_dat(circ, genelist[1:geneNum,], go$Term[1:termNum])
tiff(file="GOcircos.tiff",width = 1000,height = 1020)
GOChord(chord,
space = 0.001, #基因之间的间距
gene.order = 'logFC', #排序基因
gene.space = 0.25, #基因离圆圈距离
gene.size = 3, #
border.size = 0.1,
process.label = 7) #GO名称大小
dev.off()
# 聚类圈图
tiff(file="GOcluster.tiff",width = 1200,height = 900)
GOCluster(circ, as.character(go[1:termNum,3]))
dev.off()效果:
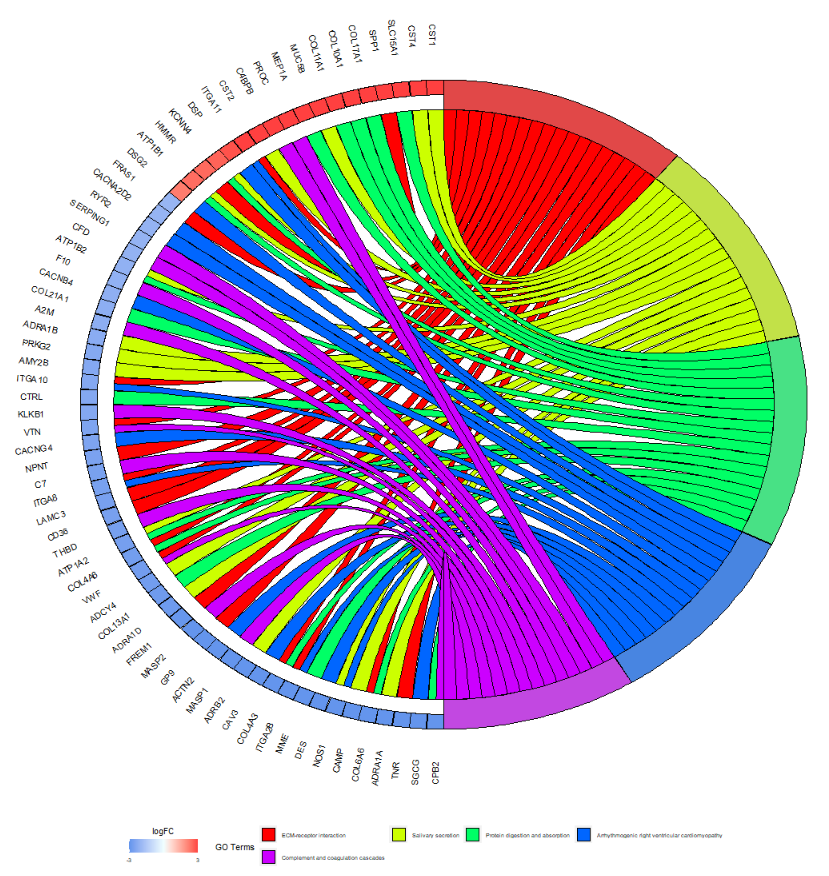
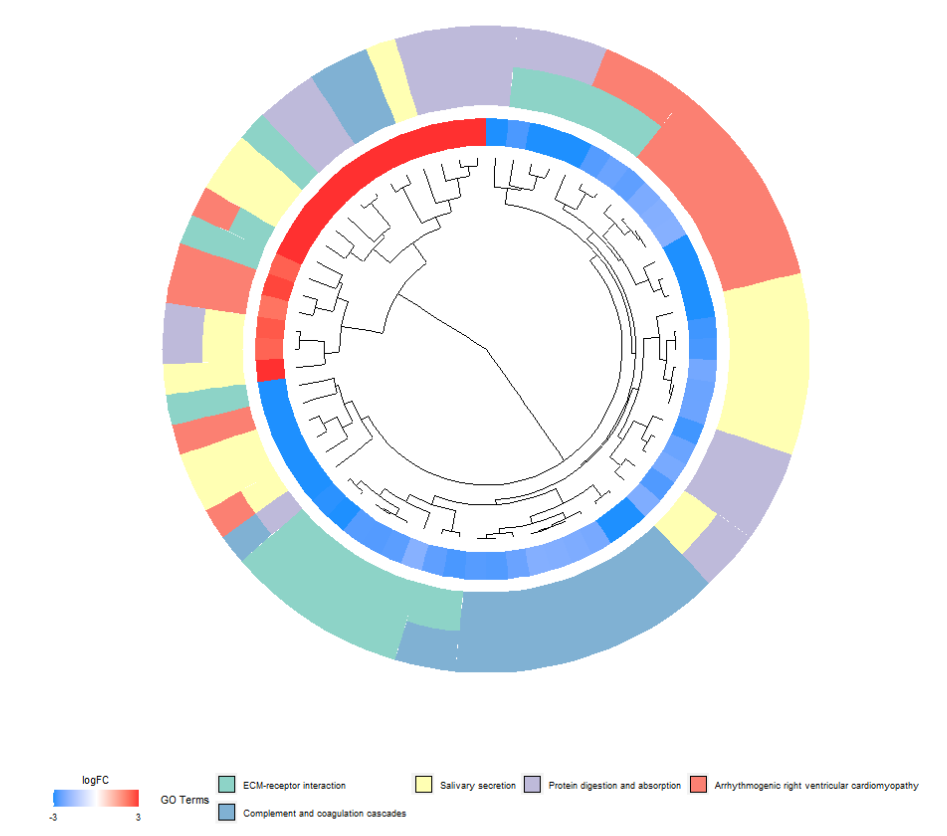
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(colorspace)) install.packages("colorspace")
if (!require(stringi)) install.packages("stringi")
if (!require(digest)) install.packages("digest")
if (!require(GOplot)) install.packages("GOplot")
if (!require(BiocManager)) install.packages("BiocManager")
# 生信的专用库
if (!require(org.Hs.eg.db)) BiocManager::install("org.Hs.eg.db")
if (!require(DOSE)) BiocManager::install("DOSE")
if (!require(clusterProfiler)) BiocManager::install("clusterProfiler")
if (!require(enrichplot)) BiocManager::install("enrichplot")
# 加载库
library(clusterProfiler)
library(org.Hs.eg.db)
library(enrichplot)
library(ggplot2)
library(GOplot)
# 过滤
pFilter=0.05
adjPfilter=0.05
rt=read.table("input.txt",sep="\t",header=T,check.names=F) #读取id.txt文件
genes=as.vector(rt[,1]) #提取基因列表
entrezIDs=mget(genes, org.Hs.egSYMBOL2EG, ifnotfound=NA) #找出基因对应id
entrezIDs=as.character(entrezIDs)
rt=cbind(rt,entrezID=entrezIDs)
rt=rt[is.na(rt[,"entrezID"])==F,] #去除id为NA的基因
gene=rt$entrezID
# KEGG富集分析
kk=enrichKEGG(gene = gene, organism = "hsa", pvalueCutoff =1, qvalueCutoff =1)
KEGG=as.data.frame(kk)
KEGG=KEGG[(KEGG$pvalue<pFilter & KEGG$p.adjust<adjPfilter),]
KEGG$geneID=as.character(sapply(KEGG$geneID,function(x)paste(rt$gene[match(strsplit(x,"/")[[1]],as.character(rt$entrezID))],collapse="/")))
write.table(KEGG,file="KEGG.txt",sep="\t",quote=F,row.names = F) #保存富集结果
# 获取KEGG信息
kegg=data.frame(Category = "ALL",ID = KEGG$ID,Term = KEGG$Description, Genes = gsub("/", ", ", KEGG$geneID), adj_pval = KEGG$p.adjust)
# 读取基因的logFC
genelist <- data.frame(ID = rt$gene, logFC = rt$logFC)
row.names(genelist)=genelist[,1]
circ <- circle_dat(kegg, genelist)
termNum = 5 #限定Term数
termNum=ifelse(nrow(kegg)<termNum,nrow(kegg),termNum)
geneNum = nrow(genelist) #限定基因数目
# 绘制圈图
chord <- chord_dat(circ, genelist[1:geneNum,], kegg$Term[1:termNum])
tiff(file="KEGGcircos.tiff",width = 1100,height = 1120)
GOChord(chord,
space = 0.001, #基因之间的间距
gene.order = 'logFC', #按照logFC对基因排序
gene.space = 0.25, #基因名跟圆圈的相对距离
gene.size = 4, #基因名字体大小
border.size = 0.1, #线条粗细
process.label = 7) #term字体大小
dev.off()
#绘制聚类圈图
tiff(file="KEGGcluster.tiff",width = 12,height = 9)
GOCluster(circ, as.character(kegg[1:termNum,3]))
dev.off()效果:
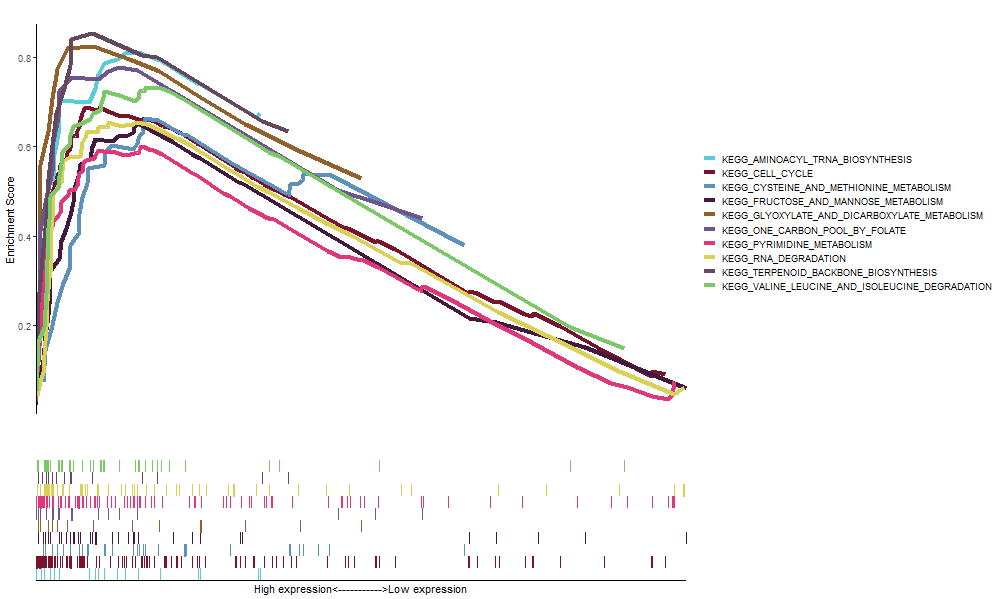
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(plyr)) install.packages("plyr")
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(grid)) install.packages("grid")
if (!require(gridExtra)) install.packages("gridExtra")
#引用
library(plyr)
library(ggplot2)
library(grid)
library(gridExtra)
files=grep(".xls",dir(),value=T) #读取目录下的所有xls文件
data = lapply(files,read.delim) #读取每个文件
names(data) = files
dataSet = ldply(data, data.frame)
dataSet$pathway = gsub(".xls","",dataSet$.id) #将文件后缀删掉
gseaCol=c("#58CDD9","#7A142C","#5D90BA","#431A3D","#91612D","#6E568C","#E0367A","#D8D155","#64495D","#7CC767","#223D6C","#D20A13","#FFD121","#088247","#11AA4D")
#富集图
pGsea=ggplot(dataSet,aes(x=RANK.IN.GENE.LIST,y=RUNNING.ES,colour=pathway,group=pathway))+
geom_line(size = 1.5) + scale_color_manual(values = gseaCol[1:nrow(dataSet)]) +
labs(x = "", y = "Enrichment Score", title = "") + scale_x_continuous(expand = c(0, 0)) +
scale_y_continuous(expand = c(0, 0),limits = c(min(dataSet$RUNNING.ES - 0.02), max(dataSet$RUNNING.ES + 0.02))) +
theme_bw() + theme(panel.grid = element_blank()) + theme(panel.border = element_blank()) + theme(axis.line = element_line(colour = "black")) + theme(axis.line.x = element_blank(),axis.ticks.x = element_blank(),axis.text.x = element_blank()) +
geom_hline(yintercept = 0) +
guides(colour = guide_legend(title = NULL)) + theme(legend.background = element_blank()) + theme(legend.key = element_blank())+theme(legend.key.size=unit(0.5,'cm'))
#基因排序图
pGene=ggplot(dataSet,aes(RANK.IN.GENE.LIST,pathway,colour=pathway))+geom_tile()+
scale_color_manual(values = gseaCol[1:nrow(dataSet)]) +
labs(x = "High expression<----------->Low expression", y = "", title = "") +
scale_x_discrete(expand = c(0, 0)) + scale_y_discrete(expand = c(0, 0)) +
theme_bw() + theme(panel.grid = element_blank()) + theme(panel.border = element_blank()) + theme(axis.line = element_line(colour = "black"))+
theme(axis.line.y = element_blank(),axis.ticks.y = element_blank(),axis.text.y = element_blank())+ guides(color=FALSE)
gGsea = ggplot_gtable(ggplot_build(pGsea))
gGene = ggplot_gtable(ggplot_build(pGene))
maxWidth = grid::unit.pmax(gGsea$widths, gGene$widths)
gGsea$widths = as.list(maxWidth)
gGene$widths = as.list(maxWidth)
# 保存图像
tiff('multipleGSEA.tiff', width=1000, height=600)
par(mar=c(5,5,2,5))
grid.arrange(arrangeGrob(gGsea,gGene,nrow=2,heights=c(.8,.3)))
dev.off()效果:
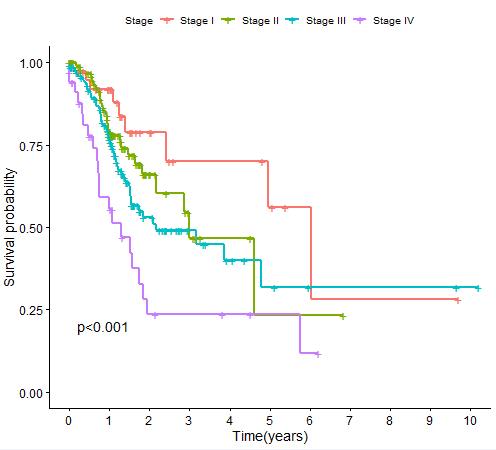
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
# 加载库
library(survival)
library(survminer)
inputFile="input.txt"
outFile="survival.tiff"
var="Stage" #用于生存分析的变量
# 读取
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
rt=rt[,c("futime","fustat",var)]
colnames(rt)[3]="Type"
groupNum=length(levels(factor(rt[,"Type"])))
# 比较组间生存差异的P值
diff=survdiff(Surv(futime, fustat) ~Type,data = rt)
pValue=1-pchisq(diff$chisq,df=(groupNum-1)) #df自由度
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf(".03f",pValue))
}
fit <- survfit(Surv(futime, fustat) ~ Type, data = rt)
# 绘制
surPlot=ggsurvplot(fit,
data=rt,
conf.int=F, #置信区间
pval=pValue,
pval.size=5,
legend.labs=levels(factor(rt[,"Type"])),
legend.title=var,
xlab="Time(years)",
break.time.by = 1,
risk.table.title="",
risk.table=F,
risk.table.height=.25)
tiff(file=outFile,width = 500,height =450)
print(surPlot)
dev.off()效果:
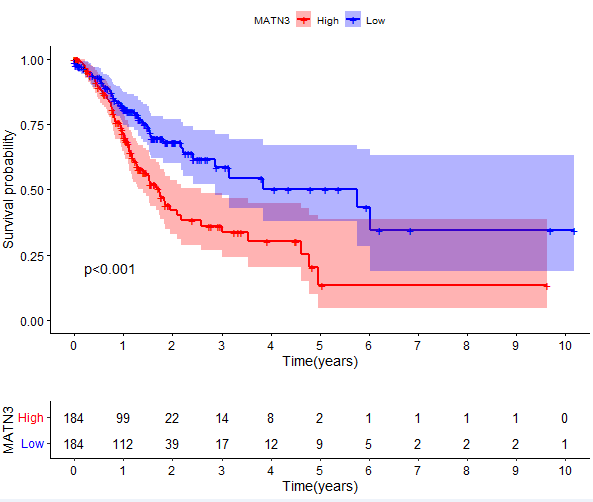
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
# 加载库
library(survival)
library(survminer)
inputFile="input.txt"
outFile="survival.tiff"
var="MATN3" #用于生存分析的变量
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
rt=rt[,c("futime","fustat",var)]
# 根据中位值,把样品分为两组
group=ifelse(rt[,3]>median(rt[,3]),"High","Low")
diff=survdiff(Surv(futime, fustat) ~group,data = rt)
pValue=1-pchisq(diff$chisq,df=1)
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf("%.03f",pValue))
}
fit <- survfit(Surv(futime, fustat) ~ group, data = rt) #剩余数目
# 绘制
surPlot=ggsurvplot(fit,
data=rt,
conf.int=TRUE,
pval=pValue,
pval.size=5,
legend.labs=c("High", "Low"),
legend.title=var,
xlab="Time(years)",
break.time.by = 1,
risk.table.title="",
palette=c("red", "blue"),
risk.table=T,
risk.table.height=.25)
tiff(file=outFile,width = 600,height =500)
print(surPlot)
dev.off()效果:
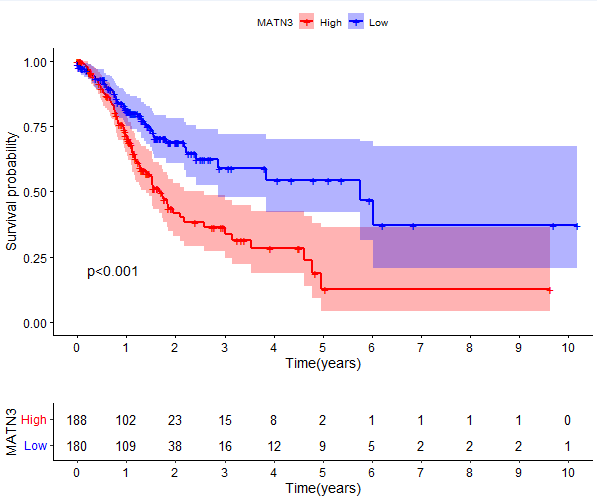
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
# 加载库
library(survival)
library(survminer)
inputFile="input.txt"
outFile="survival.tiff"
var="MATN3"
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
rt=rt[,c("futime","fustat",var)]
colnames(rt)=c("futime","fustat","var")
# 获取最优cutoff
res.cut=surv_cutpoint(rt, time = "futime", event = "fustat",variables =c("var"))
res.cut
res.cat=surv_categorize(res.cut)
fit=survfit(Surv(futime, fustat) ~var, data = res.cat)
# 比较高低表达生存差异
diff=survdiff(Surv(futime, fustat) ~var,data =res.cat)
pValue=1-pchisq(diff$chisq,df=1)
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf("%.03f",pValue))
}
# 绘制
surPlot=ggsurvplot(fit,
data=res.cat,
conf.int=TRUE,
pval=pValue,
pval.size=5,
legend.labs=c("High", "Low"),
legend.title=var,
xlab="Time(years)",
break.time.by = 1,
risk.table.title="",
palette=c("red", "blue"),
risk.table=T,
risk.table.height=.25)
tiff(file=outFile, width = 600, height =500)
print(surPlot)
dev.off()效果:
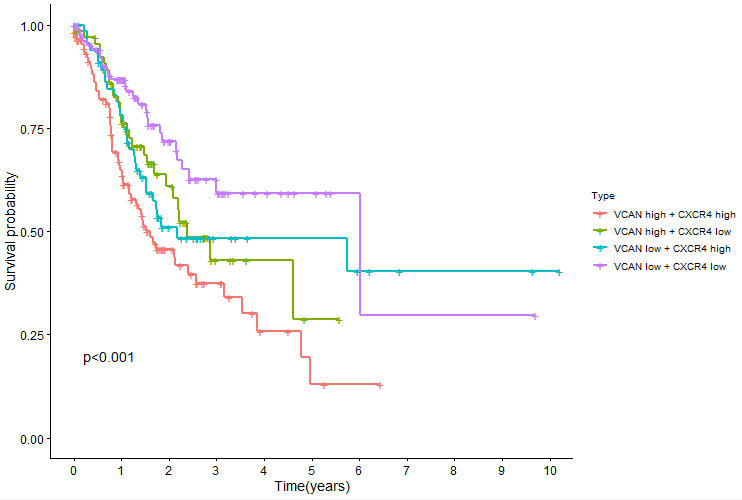
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
# 加载库
library(survival)
library(survminer)
inputFile="input.txt"
outFile="survival.tiff"
var1="VCAN" #用于生存分析的变量1
var2="CXCR4" #用于生存分析的变量2
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
#根据基因表达,对数据分组
a=ifelse(rt[,var1]<median(rt[,var1]),paste0(var1," low"),paste0(var1," high"))
b=ifelse(rt[,var2]<median(rt[,var2]),paste0(var2," low"),paste0(var2," high"))
Type=paste(a,"+",b)
rt=cbind(rt,Type)
#生存差异统计
length=length(levels(factor(Type)))
diff=survdiff(Surv(futime, fustat) ~Type,data = rt)
pValue=1-pchisq(diff$chisq,df=length-1)
if(pValue<0.001){
pValue="p<0.001"
}else{
pValue=paste0("p=",sprintf("%.03f",pValue))
}
fit <- survfit(Surv(futime, fustat) ~ Type, data = rt)
#绘制生存曲线
surPlot=ggsurvplot(fit,
data=rt,
conf.int=F, #不含置信区间
pval=pValue,
pval.size=5,
legend.labs=levels(factor(rt[,"Type"])),
legend.title="Type",
#font.legend=6,
legend = "right",
xlab="Time(years)",
break.time.by = 1,
risk.table.title="",
risk.table=F, #风险表格
risk.table.height=.25)
tiff(file=outFile,width = 750,height =500)
print(surPlot)
dev.off()效果:
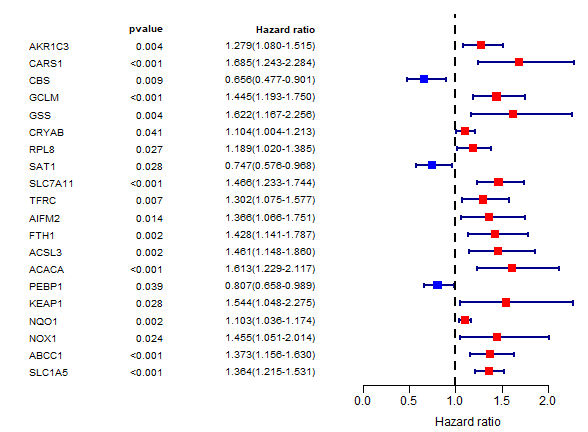
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
inputFile="input.txt"
outFile="forest.tiff"
rt=read.table(inputFile,header=T,sep="\t",row.names=1,check.names=F)
gene=rownames(rt)
hr=sprintf("%.3f",rt$"HR")
hrLow=sprintf("%.3f",rt$"HR.95L")
hrHigh=sprintf("%.3f",rt$"HR.95H")
Hazard.ratio=paste0(hr,"(",hrLow,"-",hrHigh,")")
pVal=ifelse(rt$pvalue<0.001, "<0.001", sprintf("%.3f", rt$pvalue))
# 图像格式
tiff(file=outFile, width = 600, height =450)
n=nrow(rt)
nRow=n+1
ylim=c(1,nRow)
layout(matrix(c(1,2),nc=2),width=c(3,2))
# 森林图左边的基因信息
xlim = c(0,3)
par(mar=c(4,2,1.5,1.5))
plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,xlab="",ylab="")
text.cex=0.8
text(0,n:1,gene,adj=0,cex=text.cex)
text(1.5-0.5*0.2,n:1,pVal,adj=1,cex=text.cex)
text(1.5-0.5*0.2,n+1,'pvalue',cex=text.cex,font=2,adj=1)
text(3,n:1,Hazard.ratio,adj=1,cex=text.cex)
text(3,n+1,'Hazard ratio',cex=text.cex,font=2,adj=1,)
par(mar=c(4,1,1.5,1),mgp=c(2,0.5,0))
xlim = c(0,max(as.numeric(hrLow),as.numeric(hrHigh)))
plot(1,xlim=xlim,ylim=ylim,type="n",axes=F,ylab="",xaxs="i",xlab="Hazard ratio")
arrows(as.numeric(hrLow),n:1,as.numeric(hrHigh),n:1,angle=90,code=3,length=0.03,col="darkblue",lwd=2.5)
abline(v=1,col="black",lty=2,lwd=2)
boxcolor = ifelse(as.numeric(hr) > 1, 'red', 'blue')
points(as.numeric(hr), n:1, pch = 15, col = boxcolor, cex=1.5)
axis(1)
dev.off()效果:
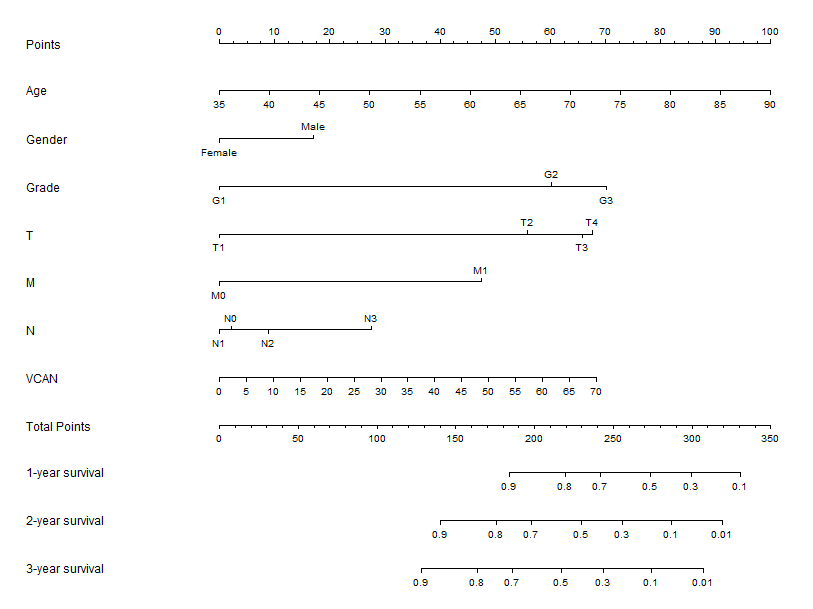
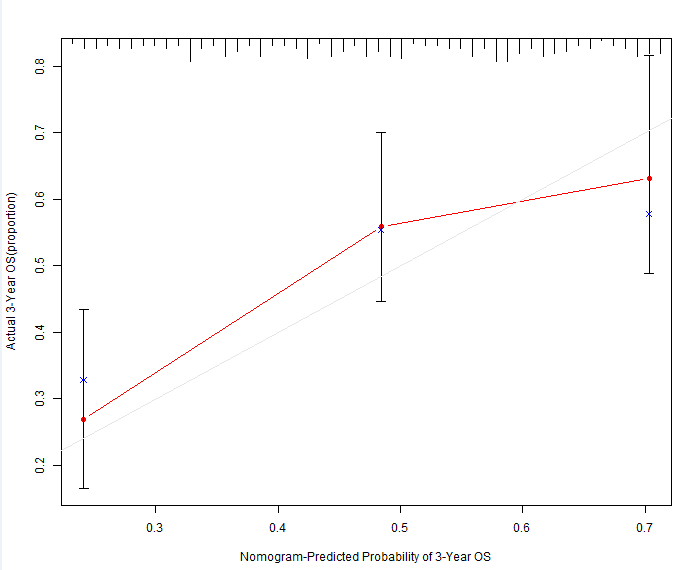
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(rms)) install.packages("rms")
# 加载库
library(rms)
inputFile="input.txt"
rt=read.table(inputFile,header=T,sep="\t",check.names=F,row.names=1)
# 数据打包
dd <- datadist(rt)
options(datadist="dd")
# 生成函数
f <- cph(Surv(futime, fustat) ~ Age+Gender+Grade+T+M+N+VCAN, x=T, y=T, surv=T, data=rt, time.inc=1)
surv <- Survival(f)
# 建立nomogram
nom <- nomogram(f, fun=list(function(x) surv(1, x), function(x) surv(2, x), function(x) surv(3, x)),
lp=F, funlabel=c("1-year survival", "2-year survival", "3-year survival"),
maxscale=100,
fun.at=c(0.99, 0.9, 0.8, 0.7, 0.5, 0.3,0.1,0.01))
# nomogram可视化
tiff(file="Nomogram.tiff",height=700,width=850)
plot(nom)
dev.off()
# calibration curve
time=3 #预测三年calibration
f <- cph(Surv(futime, fustat) ~ Age+Gender+Grade+T+M+N+VCAN, x=T, y=T, surv=T, data=rt, time.inc=time)
cal <- calibrate(f, cmethod="KM", method="boot", u=time, m=100, B=1000) #m样品数目1/3
tiff(file="calibration.tiff",height=600,width=700)
plot(cal,xlab="Nomogram-Predicted Probability of 3-Year OS",ylab="Actual 3-Year OS(proportion)",col="red",sub=F)
dev.off()效果:
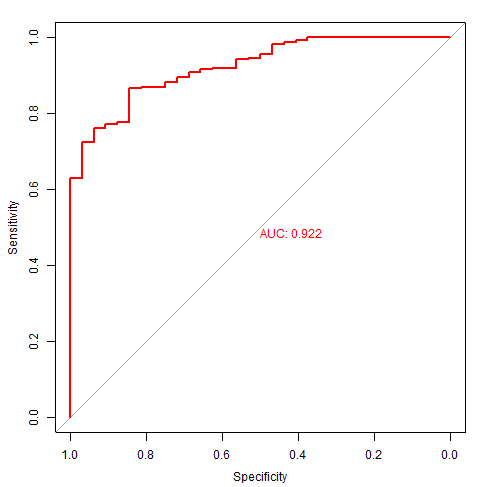
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(pROC)) install.packages("pROC")
# 加载库
library(pROC)
inputFile="input.txt"
outFile="ROC.tiff"
x="TTYH3" #用于ROC的变量
# 读入文件,整理
rt=read.table(inputFile,header=T,sep="\t",check.names=F,row.names=1)
y=colnames(rt)[1]
# 绘制
rocobj1=roc(rt[,y], as.vector(rt[,x]))
tiff(file=outFile,width=500,height=500)
plot(rocobj1, print.auc=TRUE, col="red")
dev.off()效果:
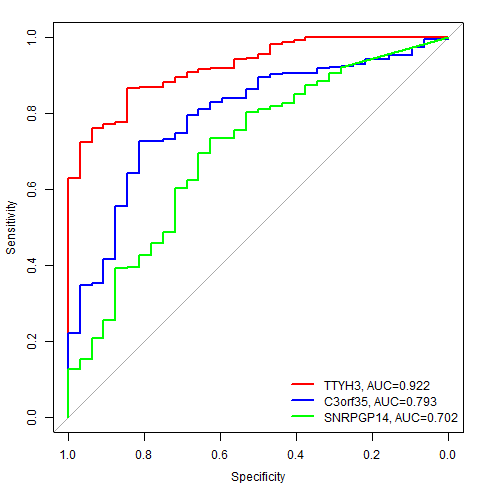
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(pROC)) install.packages("pROC")
# 加载库
library(pROC)
inputFile="input.txt"
outFile="ROC.tiff"
rt=read.table(inputFile,header=T,sep="\t",check.names=F,row.names=1)
y=colnames(rt)[1]
# 定义颜色
bioCol=c("red","blue","green","yellow")
if(ncol(rt)>4){
bioCol=rainbow(ncol(rt))}
# 绘制
tiff(file=outFile,width=500,height=500)
roc1=roc(rt[,y], as.vector(rt[,2]))
aucText=c( paste0(colnames(rt)[2],", AUC=",sprintf("%0.3f",auc(roc1))) )
plot(roc1, col=bioCol[1])
for(i in 3:ncol(rt)){
roc1=roc(rt[,y], as.vector(rt[,i]))
lines(roc1, col=bioCol[i-1])
aucText=c(aucText, paste0(colnames(rt)[i],", AUC=",sprintf("%0.3f",auc(roc1))) )
}
legend("bottomright", aucText,lwd=2,bty="n",col=bioCol[1:(ncol(rt)-1)])
dev.off()效果:
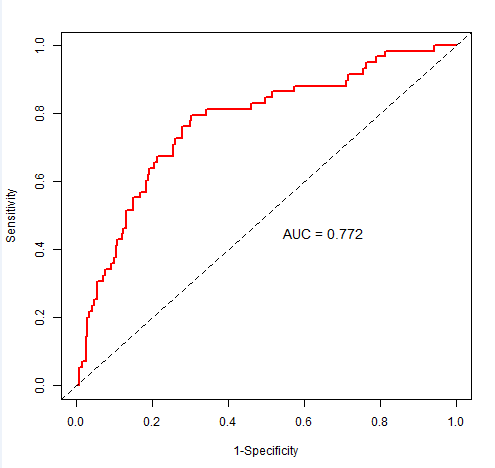
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
if (!require(timeROC)) install.packages("timeROC")
# 加载库
library(survival)
library(survminer)
library(timeROC)
inputFile="input.txt"
outFile="ROC.tiff"
var="score" #需要的变量
# 读取
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
# 绘制
ROC_rt=timeROC(T=rt$futime, delta=rt$fustat,
marker=rt[,var], cause=1,
weighting='aalen',
times=c(1), ROC=TRUE) #预测时间1年
tiff(file=outFile, width=500, height=500)
plot(ROC_rt, time=1, col='red', title=FALSE, lwd=2)
text(0.65,0.45,paste0('AUC = ',sprintf("%.03f",ROC_rt$AUC[2])),cex=1.2)
dev.off()效果:
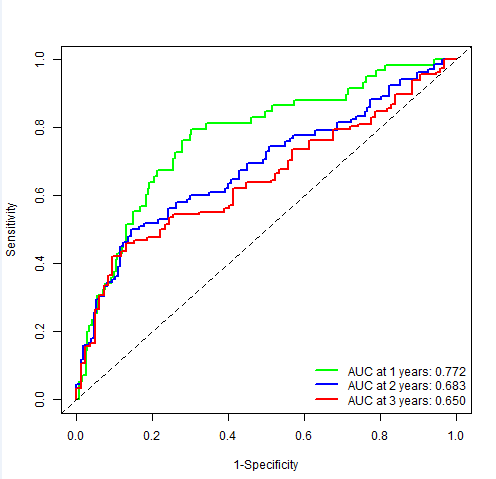
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
if (!require(timeROC)) install.packages("timeROC")
# 加载库
library(survival)
library(survminer)
library(timeROC)
inputFile="input.txt"
outFile="ROC.tiff"
var="score"
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
#绘制
ROC_rt=timeROC(T=rt$futime, delta=rt$fustat,
marker=rt[,var], cause=1,
weighting='aalen',
times=c(1,2,3), ROC=TRUE)
tiff(file=outFile,width=500,height=500)
plot(ROC_rt,time=1,col='green',title=FALSE,lwd=2)
plot(ROC_rt,time=2,col='blue',add=TRUE,title=FALSE,lwd=2)
plot(ROC_rt,time=3,col='red',add=TRUE,title=FALSE,lwd=2)
legend('bottomright',
c(paste0('AUC at 1 years: ',sprintf("%.03f",ROC_rt$AUC[1])),
paste0('AUC at 2 years: ',sprintf("%.03f",ROC_rt$AUC[2])),
paste0('AUC at 3 years: ',sprintf("%.03f",ROC_rt$AUC[3]))),
col=c("green",'blue','red'),lwd=2,bty = 'n')
dev.off()效果:
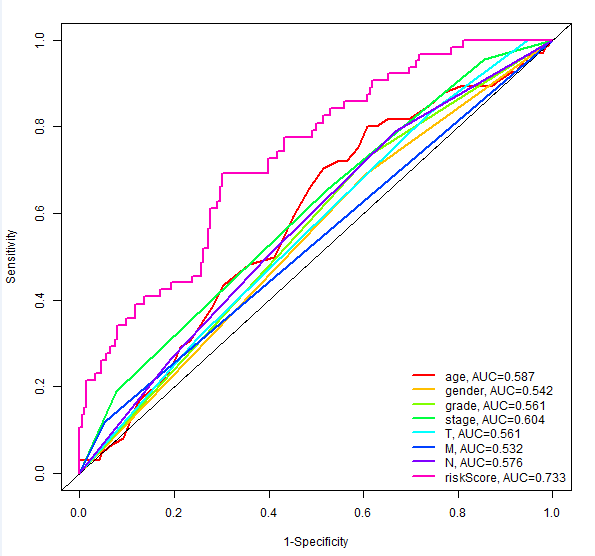
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(survival)) install.packages("survival")
if (!require(survminer)) install.packages("survminer")
if (!require(timeROC)) install.packages("timeROC")
# 加载库
library(survival)
library(survminer)
library(timeROC)
inputFile="input.txt"
outFile="ROC.tiff"
rt=read.table(inputFile, header=T, sep="\t", check.names=F, row.names=1)
# 颜色
bioCol=rainbow(ncol(rt)-2)
# 绘制
aucText=c()
tiff(file=outFile,width=600,height=600)
i=3
ROC_rt=timeROC(T=rt$futime,delta=rt$fustat,marker=rt[,i],cause=1,weighting='aalen',times=c(1),ROC=TRUE)
plot(ROC_rt,time=1,col=bioCol[i-2],title=FALSE,lwd=2)
aucText=c(paste0(colnames(rt)[i],", AUC=",sprintf("%.3f",ROC_rt$AUC[2])))
abline(0,1)
for(i in 4:ncol(rt)){
ROC_rt=timeROC(T=rt$futime,delta=rt$fustat,marker=rt[,i],cause=1,weighting='aalen',times=c(1),ROC=TRUE)
plot(ROC_rt,time=1,col=bioCol[i-2],title=FALSE,lwd=2,add=TRUE)
aucText=c(aucText,paste0(colnames(rt)[i],", AUC=",sprintf("%.3f",ROC_rt$AUC[2])))
}
legend("bottomright", aucText,lwd=2,bty="n",col=bioCol[1:(ncol(rt)-2)])
dev.off()效果:
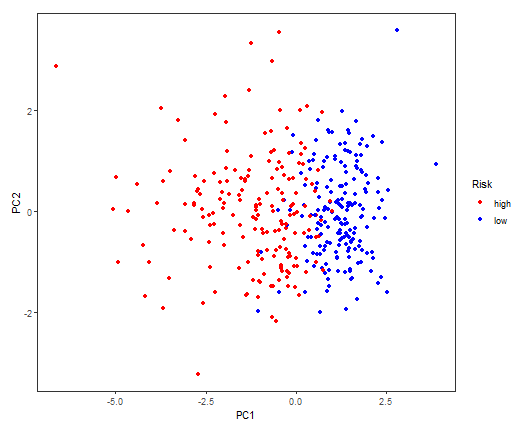
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
# 加载库
library(ggplot2)
inputFile="input.txt"
outFile="PCA.tiff"
rt=read.table(inputFile,header=T,sep="\t",check.names=F,row.names=1)
data=rt[,c(2:ncol(rt))]
Type=rt[,1]
var=colnames(rt)[1]
# PCA分析
data.pca=prcomp(data, scale. = TRUE)
pcaPredict=predict(data.pca)
PCA = data.frame(PC1=pcaPredict[,1], PC2=pcaPredict[,2], Type=Type)
# 定义颜色
col=c("red","blue")
if(length(levels(factor(Type)))>2){
col=rainbow(length(levels(factor(Type))))}
# 绘制PCA
tiff(file=outFile, height=450, width=550) #???????????ļ?
p=ggplot(data = PCA, aes(PC1, PC2)) + geom_point(aes(color = Type)) +
scale_colour_manual(name=var, values =col)+
theme_bw()+
theme(plot.margin=unit(rep(1.5,4),'lines'))+
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank())
print(p)
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(scatterplot3d)) install.packages("scatterplot3d")
# 加载库
library(scatterplot3d)
inputFile="input.txt"
outFile="PCA.tiff"
rt=read.table(inputFile,header=T,sep="\t",check.names=F,row.names=1)
data=rt[,c(2:ncol(rt))]
Type=rt[,1] #提取分组信息
var=colnames(rt)[1]
#颜色
group=levels(factor(Type))
bioCol=c("red","blue","green","yellow")
col= bioCol[match(Type,group)]
#PCA分析
data.pca=prcomp(data, scale. = TRUE)
pcaPredict=predict(data.pca)
#绘制
tiff(file=outFile, height=500, width=600)
par(oma=c(0.5,0.5,0.5,0.5))
s3d=scatterplot3d(pcaPredict[,1:3], pch = 16, color=col)
legend("top", legend =group,pch = 16, inset = -0.2, box.col="white",xpd = TRUE, horiz = TRUE,col=bioCol[1:length(group)])
dev.off()效果:
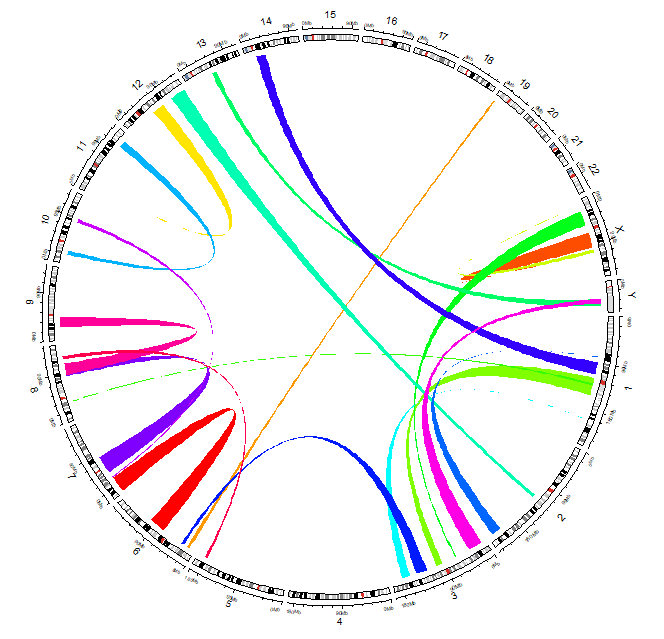
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(circlize)) install.packages("circlize")
# 加载库
library(circlize)
bedFile1="bed1.txt"
bedFile2="bed2.txt"
outFile="circos.tiff"
bed1 = read.table(bedFile1, header=T, sep="\t", check.names=F) #读取输入文件1
bed2 = read.table(bedFile2, header=T, sep="\t", check.names=F) #2
#绘制
tiff(file=outFile, width=700, height=700)
circos.par("track.height" = 0.2, cell.padding = c(0, 0, 0, 0))
circos.initializeWithIdeogram()
circos.genomicLink(bed1, bed2, col =rainbow(nrow(bed1)), border = NA)#连线
circos.clear()
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(BiocManager)) install.packages("BiocManager")
if (!require(karyoploteR)) BiocManager::install("karyoploteR")
# 加载库
library(karyoploteR)
inputFile="input.txt"
outFile="genome.tiff"
rt=read.table(inputFile, header=T, sep="\t", check.names=F)
#提取数据
data.points=makeGRangesFromDataFrame(rt)
mcols(data.points)=data.frame(y=rt[,4])
#可视化
ymin=quantile(rt[,4],0.1)
ymax=quantile(rt[,4],0.9)*2
tiff(file=outFile, width=1000, height=700)
kp=plotKaryotype("hg38", plot.type=1)
kpDataBackground(kp, data.panel=1, color="white")
kpPoints(kp, data=data.points, pch=".", col="red", ymin=ymin, ymax=ymax, cex=6)
kpAddBaseNumbers(kp, tick.dist=10000000, minor.tick.dist=1000000)
dev.off()效果:

代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(ggplot2)) install.packages("ggplot2")
if (!require(ggtree)) install.packages("ggtree")
if (!require(colorspace)) install.packages("colorspace")
# 加载库
library("ggplot2")
library("ggtree")
library("colorspace")
treFile="input.tre" #进化树文件
groupFile="group.txt" #树枝分类
outFile="tree.tiff" #输出
#读取属性文件,把属性信息保存到list
cls=list()
rt=read.table(groupFile,sep="\t",header=T)
for(i in 1:nrow(rt)){
otu=as.character(rt[i,1])
phylum=as.character(rt[i,2])
cls[[phylum]]=c(cls[[phylum]], otu)
}
phylumNames=names(cls)
phylumNum=length(phylumNames)
#读取进化树文件,和属性文件合并
tree=read.tree(treFile)
tree=groupOTU(tree, cls)
#绘制
tiff(file=outFile, width=700, height=700)
ggtree(tree,
layout="circular",
ladderize = F,
branch.length="none",
aes(color=group)) +
scale_color_manual(values=c(rainbow_hcl(phylumNum+1)),breaks=phylumNames, labels=phylumNames ) +
theme(legend.position="right") +
geom_text(aes(label=paste(" ",label,sep=""),
angle=angle+45),
size=2)
dev.off()效果:
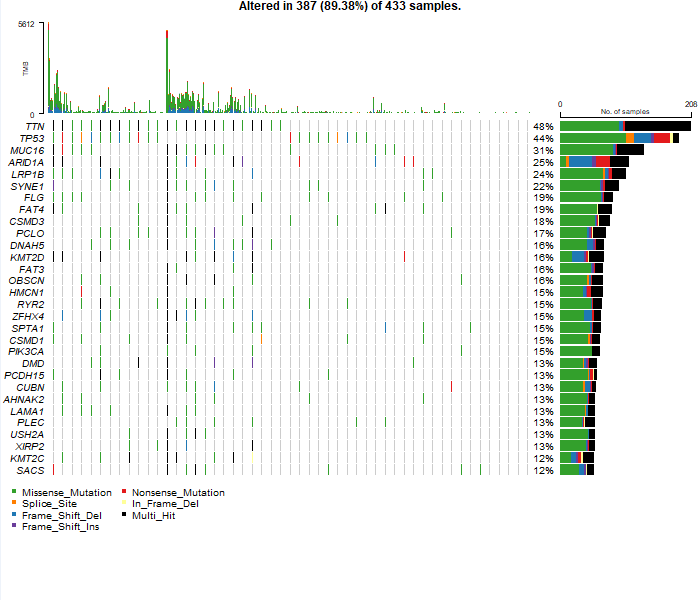
代码:
####公众号:熊大学习社####
# 手动设置工作目录为代码和数据所在文件夹
# 步骤方法:点菜单栏“session”->"Set Work Directory"->"Choose Directory",选择代码和数据所在文件夹即可
# 查看工作目录
getwd()
# 安装库
if (!require(BiocManager)) install.packages("BiocManager")
if (!require(maftools)) BiocManager::install("maftools")
# 加载库
library(maftools)
inputFile='input.maf'
outFile="waterfall.tiff"
maf=read.maf(maf = inputFile )
#绘制
tiff(file=outFile,width=700,height=600)
oncoplot(maf = maf, top = 30, fontSize = 0.8 ,showTumorSampleBarcodes = F )
dev.off()(1)学习资料,包括医学SCI绘图-基于R课程的50+种学术图片的代码和数据。关注公众号“熊大学习社”,回复“scih1214”,获取资料链接,代码在文章中。50+种学术图片的代码和数据资料在文章最下面,付费后可见。
谢谢您的支持,我们坚持学以致用、高效学习、质量服务,做好有质量的分享。
我们提供专业的SCI论文指导、数据分析、翻译润色、写作辅导等服务,了解请扫码加客服。
关注B站熊大学习社,公众号熊大学习社、诺维之舟。您的一键三连是我最大的动力。